
With over 100 nucleoside analogues already in our catalog, it becomes more and more difficult to find new and exciting nucleoside products. Of course, the most accessible modified nucleosides have already been produced as phosphoramidites. In the last year, we have continued to target some of our favorite applications - DNA damage and repair, modulation of hybridization, fluorescent products, and novel labelling procedures. In conjunction with our chemistry collaborators at Berry and Associates, we have evaluated more than a dozen new products in the last year. Unfortunately, the majority did not make the cut, but we hope the set that follows will find an interested audience.
Thymine glycol (5,6-dihydroxy-5,6-dihydrothymine) (1) is formed when thymine is subjected to oxidative stress, including ionizing radiation. Oxidation of the 5,6 double bond of Thymidine generates two chiral centers at C5 and C6. The cis-5R,6S form (1) is generated as the predominant product along with the other diastereomer, the cis-5S,6R form (2). The presence of thymidine glycol in DNA has significant biological consequences and many organisms possess specific repair enzymes for the excision of this lesion. Previously, oligonucleotides containing thymidine glycol were formed by subjecting them to post synthetic oxidation using potassium permanganate or osmium tetroxide. Yields of the desired thymidine glycol lesion were low, specificity was poor and the desired products were difficult to isolate. Despite the biological significance of this mutation, a procedure for incorporating this oxidatively damaged monomer into oligonucleotides was only recently described.1
We are happy to introduce Thymidine Glycol (Tg) CE Phosphoramidite (3) with the glycol protected with a pair of t-butyl-dimethylsilyl (TBDMS) protecting groups. Using this monomer and phosphoramidites from our UltraMild group of products, Pac-dA, Ac-dC, and iPr-Pac-dG, oligonucleotide synthesis proceeds routinely. UltraMild cleavage and deprotection using ammonium hydroxide at room temperature for 2 hours gave a clean oligonucleotide product containing thymidine glycol still protected with TBDMS groups.
Our initial attempts to remove the TBDMS groups with t-butylammonium fluoride in tetrahydrofuran (TBAF) or triethylamine trihydrofluoride (TEA.3HF) overnight at room temperature generated products that looked very good by reverse phase HPLC, but analysis by ion-exchange HPLC revealed multiple species, albeit with a major component. Electrospray mass spectro-scopic analysis of the products revealed that the TBAF product included lower molecular weight products, indicating decomposition of the deprotected thymidine glycol, while the TEA.3HF product had higher molecular weight species, indicating incomplete desilylation. In an attempt to mediate the TBAF reaction, the TBAF solution was first dried over activated molecular sieves.2 The deprotection of a simple 12 mer overnight at room temperature with dry TBAF again gave an excellent product by RP HPLC. Ion exchange HPLC (Figure 1 - TBAF) revealed a much purer product and MALDI TOF MS analysis showed that the product was predominantly the correct dihydrothymine structure with some 5-methyl-5-hydroxy-hydantoin decomposition product (6) also present. However, the best result was achieved using TEA.3HF at 40°C overnight. RP HPLC showed a pure product while ion-exchange HPLC (Figure 1 - TEA.3HF) and MS data indicated that the product was essentially pure.
 |
 |
TBAF Product |
TEA.3HF Product |
It has been reported3 that epimerization at the 6-position of thymine glycol can occur, so we must assume that the products from oligonucleotide deprotection are equilibrium mixtures of diastereomers. Recently, Iwai4 and Wang5 have published further data indicating that the equilibrium after oligonucleotide synthesis is approximately 87% cis-5R,6S (1) to 13% trans-5R,6R (4).
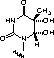





Thymidine Glycol CE Phosphoramidite (10-1096)
In the last Glen Report, we introduced 8-Amino-dA (7) and we now offer its partner 8-Amino-dG (8). 8-Amino-purines are especially interesting for their properties in triple helices. The additional amino group is effective in increasing the stability of the triple helix by the addition of a Hoogsteen purine-pyrimidine hydrogen bond, as well as integrating an amino group in the "spine of hydration" in the minor-major groove of the triple helix.6 In addition to its use in triplex oligos, 8-Amino-dG will find use in mutagenesis studies. 8-Amino-G (9) is formed along with 8-oxo-G (10), as the major mutagenic lesions formed in DNA damage caused by 2-nitropropane. 2-Nitropropane is an industrial solvent and a component of paints, dyes and varnishes, and is also present in cigarette smoke.7
The use of 8-Amino-dG in oligonucleotide synthesis is straightforward, requiring no changes from regular procedures, with the exception of the addition of 2-mercaptoethanol to the cleavage and deprotection solutions to avoid further oxidative damage.




8-Amino-dA-CE Phosphoramidite (10-1086)
8-Amino-dG-CE Phosphoramidite (10-1079)
We also described in the previous Glen Report the use of 2-F-dI (11) as a convertible nucleoside for the preparation of 2'-dG derivatives following the displacement of the 2-fluorine by primary amines.8 Following an analogous procedure, 2-F-dA (12) can be used as a convertible nucleoside to form 2-amino-dA derivatives. The fluorine in 2-F-dA is much more resistant to displacement than in 2-F-dI. However, we have successfully displaced the fluorine with dansyl cadaverine on solid phase and with methylamine in solution. Work is continuing to optimize the displacement reaction and the information on our web site will give the most up-to-date procedures.


2-F-dA-CE Phosphoramidite (10-1087) has been discontinued.
2-F-dI-CE Phosphoramidite (10-1082)
Abasic sites are produced in DNA by a variety of mechanisms, including oxidative damage, and depurination or depyrimidination by chemical and enzymatic means. DNA researchers are well served because of the availability of precursors to produce an abasic site in oligos, including our dSpacer (13)9,10, which leads to a reduced and base-stable analogue of the true abasic site and Abasic Phosphoramidite (14),used to form a true abasic site.11 Abasic sites in RNA are not so easily generated because of the greater stability of RNA to depurination, but the growing interest in RNA would seem to justify the introduction of an abasic ribose analogue for incorporation into RNA.
Abasic sites do indeed have an effect on RNA structure and activity. An example has been described using the hairpin ribozyme, which catalyzes a phosphodiester cleavage reaction. When abasic sites are introduced in various positions of the ribozyme core, ribozyme activity is greatly reduced. Interestingly, ribozyme activity could be rescued at least partially by the addition of nucleobases and the relative ability of the nucleobases to restore ribozyme activity could be used to probe ribozyme function and structure.12
We are happy to introduce rSpacer (15), the ribo-equivalent of dSpacer. For 2'-protection, we have chosen the TOM protecting group, which will make this novel monomer compatible with monomers with TBDMS or TOM13 protecting groups.

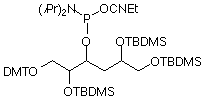

Abasic Phosphoramidite (dR Precursor) has been discontined. Please see Abasic II Phosphoramidite.
rSpacer CE Phosphoramidite has been discontined. Please see rSpacer TBDMS CE Phosphoramidite.
Abasic II Phosphoramidite (10-1927)
dSpacer CE Phosphoramidite (10-1914)
rSpacer TBDMS CE Phosphoramidite (10-3915)
We are delighted to introduce 2'-selenomethyl-U CE Phosphoramidite, the initial product derived from our association with Dr. Zhen Huang from Brooklyn College. Dr. Huang has kindly provided us with some notes on applications of 2'-Se-Me-U for X-ray crystallography. His review is on the opposite page.
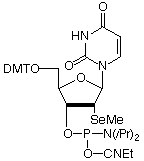
2'-Se-Me-U-CE Phosphoramidite has been discontinued
Usually, when we introduce a new product at Glen Research, we have good reasons to believe that it contains the most likely structural features for success. In the case of Amino-Modifier C6 dA, (16) (Figure 1), we already know that the structure is not optimal, in that attachment of groups to the 8 position of dA will destabilize the base pair to T. A better strategy is to attach the group at the 7-position of a 7-deaza-dA and probably the best strategy is to use 7-deaza-8-aza-dA, which also has the same electronic attributes as dA. However, these two options will yield a product which is enormously difficult to synthesize and, therefore, very expensive. Our tests with oligos containing Amino-Modifier C6 dA do indeed indicate that duplexes are destabilized by 2°C per insertion. However, Amino-Modifier C6 dA still codes specifically as dA. Although possibly not ideal, we feel that Amino-Modifier C6 dA offers a reasonable combination of performance and price.
