Glen Report 21.15: Deprotection - Volume 2 - RNA Deprotection
Introduction
| Temperature | Time | |
|---|---|---|
| RNA | ||
| AMA | RT | 20min. |
| NH4OH/EtOH (3:1) | RT | 2h |
| DNA | ||
| AMA | RT | 10min. |
| NH4OH/EtOH (3:1) | RT | 1h |
In the previous article in this series on Deprotection (Glen Report 20.2), we focused on the absolute necessity to "Deprotect to Completion" while following our mandate to "Do No Harm". In this article, we focus our attention on RNA Deprotection. Again, this is not a comprehensive review of the topic. Rather, we are attempting to offer a unified deprotection strategy that is simple to follow for newcomers to the mysterious art of RNA synthesis, while producing pure, active RNA oligos with the minimum of fuss. Where appropriate, we will mention other suitable techniques but by reference only. In the meantime, our detailed technical bulletins for RNA deprotection have also been updated and can be found on our web site by following these links:
TBDMS-Protected RNA: http://www.glenresearch.com/Technical/TB_RNA_TBDMS_Deprotection.pdf
TOM-Protected RNA: http://www.glenresearch.com/Technical/TB_RNA_TOM_Deprotection.pdf
Deprotection of RNA and chimeric DNA/RNA oligonucleotides is unique due to the requirement to retain the 2’ protecting group during cleavage, phosphate deprotection, and base deprotection. Only after the oligo is cleaved from the support, the cyanoethyl groups removed from the backbone, and bases fully deprotected, can you complete the 2’ deprotection step to yield fully functional RNA. However, we must focus on our mandate - First, Do No Harm.
First, Do No Harm
As with DNA, the modifiers or dyes present in an RNA oligonucleotide will largely dictate the types of RNA phosphoramidites required and thus, the deprotection conditions. For your consideration, we offer three types of RNA monomers, which we will describe briefly below:
TOM-Protected RNA Phosphoramidites
These monomers exhibit high coupling efficiency and are especially useful in high throughput situations since they perform better in situations where moisture control is not perfect. The high coupling efficiency allows very long RNA oligos (>75mer) to be prepared. These monomers are compatible with high speed deprotection techniques using methylamine.
TBDMS-Protected RNA Phosphoramidites
These are our workhorse monomers with an excellent cost / performance ratio. They are also compatible with high speed deprotection techniques using methylamine.
UltraMild RNA Phosphoramidites
Many minor RNA monomers, modifiers and dyes are not compatible with aggressive deprotection techniques and these UltraMild monomers will allow much milder deprotection conditions.
Any downstream purification requirements will also impact the proper handling of the RNA throughout the deprotection process. For example, DMT-on purification, e.g., Glen-Pak RNA, has become increasingly popular for the purification of RNA oligos, especially siRNA, so all deprotection schemes must leave the DMT group intact to allow purification to take place.
Cleavage
For RNA oligos we do not routinely use a separate cleavage step. By exposing the support to the full deprotection conditions, we feel that maximum yield of product in solution is achieved. Any dissolved silica will be lost in the further deprotection steps required for RNA oligos. Nevertheless, we show the recommended cleavage times for various deprotection solutions for both DNA and RNA supports in Table 1.
Deprotect to Completion
Volume 2: RNA - Deprotect to Completion
- Do I have very special components in my oligo or not?
TOM/TBDMS vs UltraMild - Do I have one or many oligos to treat?
TOM vs TBDMS - Do I need/want to purify my oligo after deprotection or not?
Precipitation vs Glen-Pak
| Temperature | Time | |
|---|---|---|
| AMA | 65°C | 10min. |
| NH4OH/EtOH (3:1) | RT | 4h |
Base Deprotection
In this article, we will focus on regular base deprotection using ammonium hydroxide/methylamine (AMA) at elevated temperature, which we have shown to be optimal for both TOM-protected and TBDMS-protected RNA, and UltraMild deprotection using ammonium hydroxide/ethanol (3:1) at room temperature for oligos containing base labile groups. It must be stressed that all of these schemes require the use of Ac-protected C monomers.1,2 We have previously3 recommended ethanolic methylamine/aqueous methylamine (1:1) (EMAM) for deprotecting TOM-protected RNA. This deprotection scheme is preferred for long RNA oligos but is not necessary for regular RNA oligos. Tables 2 shows the temperatures and times for regular RNA and UltraMild RNA deprotection.
After deprotection as above, decant the supernatant liquid from the support and evaporate to dryness. If the DMT protection has been retained for purification purposes, the solution should be evaporated using a stream of nitrogen or compressed air to avoid any loss of the DMT group. Sterile, RNase-free conditions must be maintained from this point onwards.
2’ Deprotection
In the past, we have described several schemes for removing the silyl protecting groups from the 2’-hydroxyl group. t-Butylammonium fluoride in THF (TBAF) has been used extensively for this purpose,4 as has neat triethylamine trihydrofluoride (TEA.3HF). TEA.3HF based cocktails have become much more commonly used and are compatible with both precipitation and cartridge-based downstream processing methodologies.5-7 Various additives such as triethylamine (TEA) buffer the neat TEA.3HF used in the original methods, which tended to both remove DMT and depurinate dA sites in chimeric oligos. These cocktails also function well with all three types of RNA monomers available in the Glen Research catalog. In addition, it must be noted that TBAF is not compatible with the Glen-Pak RNA purification process.
Butanol Precipitation
To complete the 2’ deprotection for a DMT-off RNA, fully re-dissolve the oligo in anhydrous DMSO. Remember to avoid glass and use a sterile or RNase free, polypropylene, o-ring capped tube for this reaction. If necessary, heat the oligo at 65°C for about 5 minutes to get the oligo fully into solution. Add TEA.3HF, mix well and heat to 65°C for 2.5 hours. Cool the solution and desalt the oligo via butanol precipitation.
DMT-on Purification
The 2’ deprotection of DMT-On RNA is slightly different due to the addition of TEA to the cocktail, thus aiding in retention of the DMT. The deprotected RNA is then quenched and immediately purified on a Glen-Pak RNA cartridge.
Fully re-dissolve the RNA in anhydrous DMSO and if necessary, heat the oligo at 65°C for about 5 minutes to get the oligo fully into solution. Add TEA to the DMSO/RNA solution and mix gently. Follow this with TEA.3HF, mix well and heat to 65°C for 2.5 hours. Instead of quenching the cocktail by the addition of butanol, add 1.75mL of RNA Quenching Buffer (60-4120-XX) to the reaction. The sample is now ready for Glen-Pak RNA cartridge purification.
Glen-Pak Purification
Glen-Pak RNA purification cartridges can purify from 40 nmole to 1.0 µmole scale syntheses in one load and are supplied in two formats; one for vacuum manifolds and another for use with a disposable syringe. Glen-Pak purified RNA oligos routinely show purities of between 90 and 95% and yields in the 50 to 80 OD range for a 1.0 micromole synthesis. Once the 2’ deprotection solution is quenched, immediately load the 2mL solution on a properly prepared Glen-Pak RNA cartridge and follow the Glen-Pak RNA procedure. At the end of the procedure, the oligo is eluted in 1.0mL RNase Free 1M ammonium bicarbonate/30% Acetonitrile and lyophilized to dryness. Ammonium bicarbonate is a volatile salt, but a second drying step from RNase free water may be required to remove excess bicarbonate.
RNA Analysis
Accurate analysis of Glen-Pak purified RNA by Ion Exchange HPLC using more traditional buffer systems can be hampered by formation of secondary structures. The use of a sodium perchlorate buffer system as well as heat should denature most oligoribonucleotides. Figure 1 shows the Ion Exchange HPLC analysis of a Glen-Pak purified, 21-mer siRNA.
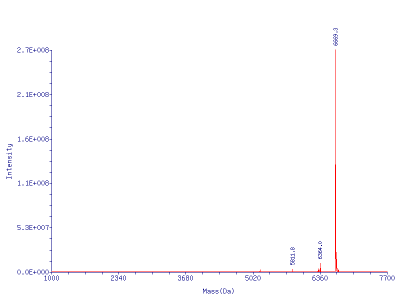
Another way to avoid secondary structure issues and obtain proper identity determination of oligoribonucleotides during purity analysis is Electrospray Mass Spectroscopy (ESI MS). Figure 1 also shows the results of an ESI MS analysis of a Glen-Pak purified 21-mer siRNA.
Further details for both DMT-On and DMT-Off 2’ RNA deprotection methods, suggested reagents, Glen-Pak Purification and downstream processing can be found in our Glen Report 19.2 (http://www.glenresearch.com//GlenReports/GR19-22.html) and the technical bulletins above.
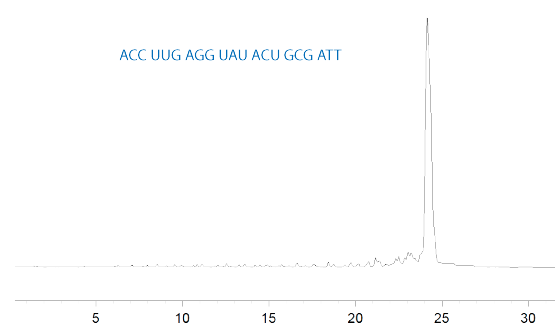
B- 600mM NaClO4, 25mM Tris-HCl, 20% Acetonitrile, pH 7.4; Gradient: 0-10% Buffer B over 30 minutes; Flow 1mL/min.
Summary
RNA synthesis is much more challenging than DNA synthesis but, as these notes indicate, it is not prohibitive. The procedures described above can be used for generating siRNA oligos of high purity on any in-house synthesizer, as demonstrated by the chromatographic and mass spec data. There is no need to go to a specialist custom oligo service for these oligos. However, the synthesis of RNA oligos >50mer in length remains challenging, but not from the aspect of synthesis, deprotection and purification. The challenge comes from the secondary structure exhibited by RNA, which makes analysis and purity determination very difficult. As interest in modified and labelled RNA oligos continues to increase, we expect to see increasing use of UltraMild techniques.
References:
- M.P. Reddy, F. Farooqui, and N.B. Hanna, Tetrahedron Letters, 1995, 36, 8929-8932.
- F. Wincott, et al., Nucleic Acids Res., 1995, 23, 2677-2684.
- S. Pitsch, P.A. Weiss, L. Jenny, A. Stutz, and X.L. Wu, Helv Chim Acta, 2001, 84, 3773-3795.
- T. Wu, K.K. Ogilvie, and R.T. Pon, Nucleic Acids Res., 1989, 17, 3501.
- E. Westman, and R. Stromberg, Nucleic Acids Res., 1994, 22, 2430-2431.
- B. Sproat, et al., Nucleosides and Nucleotides, 1995, 14, 255-273.
- D. Gasparutto, et al., Nucleic Acids Res., 1992, 20, 5159-66.