Glen Report 9.17: Cyclic Oligonucleotides
Among the many ways to stabilize oligonucleotides to exonuclease enzymes is the formation of cyclic oligonucleotides in which no terminus is left for the digestion process to commence. Cyclic oligonucleotides would clearly be of interest to antisense researchers for this property alone. In addition, properly designed cyclic structures can be used to form interesting duplex structures similar to the dumbbells and hairpin loops formed in viruses. A variety of triplex structures with the third strand without and within the cyclic oligonucleotide could also be envisaged. There are clearly many reasons to study cyclic oligonucleotides and several ways to prepare them. An excellent review1 of cyclic oligonucleotides was published earlier this year.
Preparation of Cyclic Oligonucleotides
There is no universal procedure, as yet, for the preparation of cyclic oligonucleotides, regardless of length. Short oligonucleotide circles have been prepared by chemical cyclization of protected oligonucleotides in solution2 or on solid phase.3 Since the likelihood of the ends meeting successfully decreases with oligonucleotide length, this method is most suitable for short sequences. For long sequences, a DNA template is used to line up the ends which are then joined chemically or ligated enzymatically.4,5 Therefore until now, medium-sized cyclic oligonucleotides (10- to 30-mers) have been the most difficult to prepare.
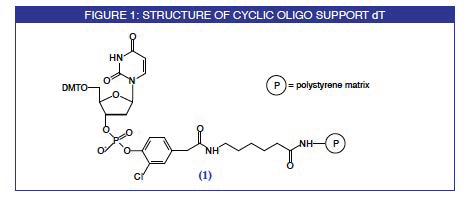
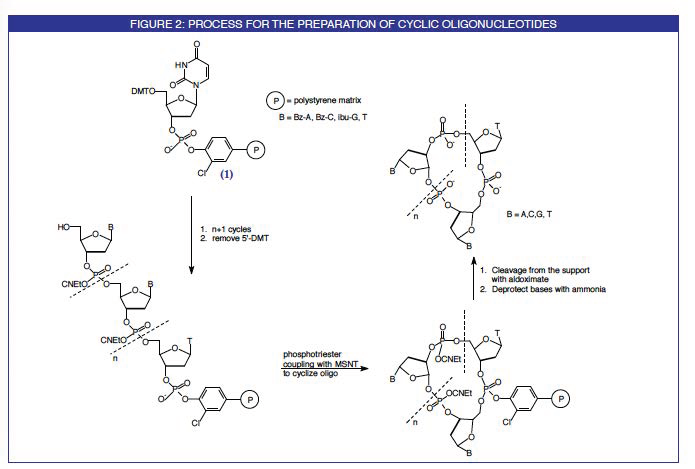
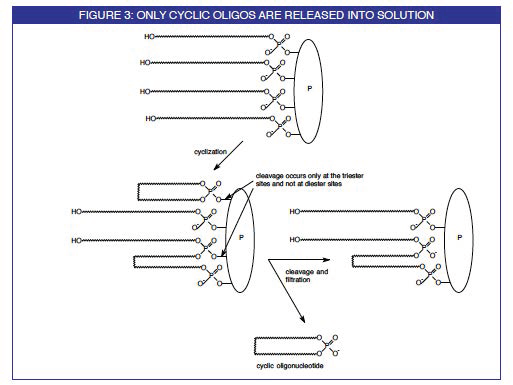
A group from the University of Barcelona has reported a simple, general and effective method for the synthesis of small- to medium-sized cyclic oligodeoxyribonucleotides. The elongation of the oligonucleotide chain is carried out on a novel support (1), Figure 1, using standard phosphoramidite chemistry. The cyclization reaction is then also carried out on the solid support using a phosphotriester coupling procedure. These steps are detailed in Figure 2. On final deprotection with ammonium hydroxide, virtually the only product which comes into solution is the desired cyclic oligonucleotide for reasons depicted in Figure 3.
Support Preparation
3-Chloro-4-hydroxyphenylacetic acid is used to anchor the oligo-nucleotide chain to the solid matrix. The reaction between its 2,4,5-trichloro-phenyl ester derivative and T-CE Phosphoramidite in the presence of tetrazole, followed by oxidation, provides the nucleotide-linker which is anchored to an amino-derivatized resin. The cyanoethyl protecting group is removed to afford the support (1). This unusual nucleotide support is offered by Glen Research under license from the University of Barcelona. Initially, we will offer only the T support since the synthesis of the cyclic oligonucleotide can commence from any T residue in the circle.
Cyclic Oligonucleotide Synthesis
From this T support, chain elongation takes place by standard phosphoramidite chemistry. After the chain assembly, removal of the 5'-DMT group allows the regioselective condensation of the 5'-terminal OH and the 3'-phosphate using 1-mesitylene-sulfonyl-3-nitro-1,2,4-triazole (O.1M in pyridine, 12-24 hours) as the cyclization reagent. Finally, the cyclic oligonucleotide is obtained after the product is cleaved from the solid support with TMG syn-pyridine-2-aldoximate (O.1M in dioxane/water, 8-16 hours) and the nucleobases are deprotected with ammonium hydroxide. After gel-filtration the crude cyclic oligonucleotides are analyzed, purified and characterized by usual procedures.
Using these strategies, cyclic oligonucleotides within the range of 2- to 30-mer have been prepared, containing all the nucleobases and without sequence restrictions. Yields of crude products depend on the size of the cyclic structure (up to 50% for the smallest cycles, 10% or less for the largest ones). However, highly pure crude cyclic oligonucleotides are obtained (typically >90% by HPLC). This fact illustrates the power of this solid-phase synthetic method, since during the cleavage step the non-cyclized product and other impurities remain anchored to the support through a phosphate diester bond, whereas the phosphate triester-linked cyclic molecule is removed from the resin, as illustrated in Figure 3.
We thank Professor Enrique Pedroso, University of Barcelona for allowing us to abstract part of the information he presented at the Nucleosides and Nucleotides Round Table in September, 1996.
References
(1) E.T. Kool, Annual Review of Biophysics and Biomolecular Structure, 1996, 25, 1-28 and references cited therein.
(2) M.V. Rao and C.B. Reese, Nucleic Acids Res., 1989,17, 8221-8239.
(3) L. De Napoli, A. Messere, D. Montesarchio, G. Piccialli, C. Santacroce, and G.M. Bonora, Nucleosides and Nucleotides, 1993,12, 21-30.
(4) G.W. Ashley and D.M. Kushlan, Biochemistry, 1991, 30, 2927-2933.
(5) G. Prakash and E.T. Kool, J. Amer. Chem. Soc., 1992,114, 3523-3527.
Note:
dT Nucleotide PS has been discountinued.