Glen Report 11.14: The Use of Puromycin CPG in Combinatorial Chemistry
New methods in the field of combinatorial chemistry make it possible to synthesize and screen large libraries of synthetic molecules for biological activity. Classically, well-defined molecules were systematically modified and the product compounds analyzed for improved biological activity. Newer combinatorial chemistry methods allow a chemist to synthesize a large population of similar compounds and then select for biological activity. (For a review1 of issues concerning combinatorial chemistry see the February 1996 issue of C&E News.) In order for the new combinatorial chemistry to be an effective tool in evaluating structure-activity relationships three fundamental criteria must be met:
- A technique must be developed to synthesize a random series of structures.
- A high-throughput screening assay must be available to evaluate the large number of synthesized molecules.
- A method must be developed to capture and analyze the structure of the selected compounds.
Random Structure Generation
Combinatorial chemistry is well suited to studying peptides and oligonucleotides. Libraries of these compounds can be easily synthesized using solid-phase chemistry. Sequence degeneracy can be incorporated during the synthesis using either split synthesis or parallel synthesis.
1. In the split synthesis approach, the solid support is divided into portions prior to each coupling step. A different synthon is then coupled to each portion. All portions are recombined after coupling and the synthesis cycle is completed. This "split and mix" approach has the advantage of yielding a unique sequence on each support bead and variability in synthon reactivity can be corrected by varying the coupling conditions. Peptide synthesis, where variations in reactivity between amino acid synthons are significant, requires the "split and mix" approach. Oligonucleotide monomers on the other hand have similar reactivity. Degeneracy can be incorporated into oligonucleotides simply by using equimolar mixes of amidites at each coupling step. Of course, this variation does not yield one sequence per bead.
2. In the parallel synthesis approach, different compounds are synthesized in multiple reaction vessels. Parallel synthesis has been performed using microtiter plates as well as in specific locations on solid surfaces that are reactive, such as the DNA chip technology developed by Affymetrix.
High-Throughput Screening
Following random structure generation, there must be a method to isolate structures that exhibit enhanced activity. Protein-binding and enzyme-activity assays are common methods that have been used to select for active structures from the library of synthesized compounds.
Capture and Analysis
One of the most challenging requirements associated with combinatorial chemistry is the recovery of sequence information of the oligonucleotide or peptide selected by the screening assay. A recent paper2 describes a novel approach to this challenge. The authors have developed a method to generate a fusion product between mRNA and the polypeptide it encodes using in vitro translation of synthetic RNAs 3'-labeled with puromycin, an antibiotic that mimics transfer RNA. Puromycin binds in the ribosome's A site, forms a peptide bond with the growing peptide chain, and blocks further peptide elongation. By linking puromycin to mRNA, a peptide-RNA fusion product results from the translation of the message linking the encoding mRNA with its peptide product.
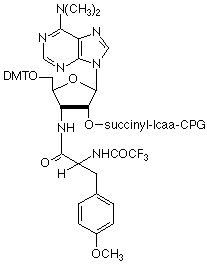
Exploiting this observation, a new support was created by attaching protected puromycin to controlled pore glass (Puromycin-CPG) (Figure 1). This support was then used to synthesize d(A27CC)-puromycin to which various mRNA sequences were then ligated. The mRNA sequence information was then translated in a reticulocyte lysate system. As the ribosome reached the poly-dA sequence, translation was stalled. Puromycin entered the ribosome A site and a peptide bond formed between the C-terminal of the synthesized peptide and the RNA encoding the peptide structure. The poly-dA sequence serves two purposes, first it stalls the ribosome thereby allowing puromycin to enter the A site and second it acts as a future capture site for oligo-dT-biotin.
Method Verification
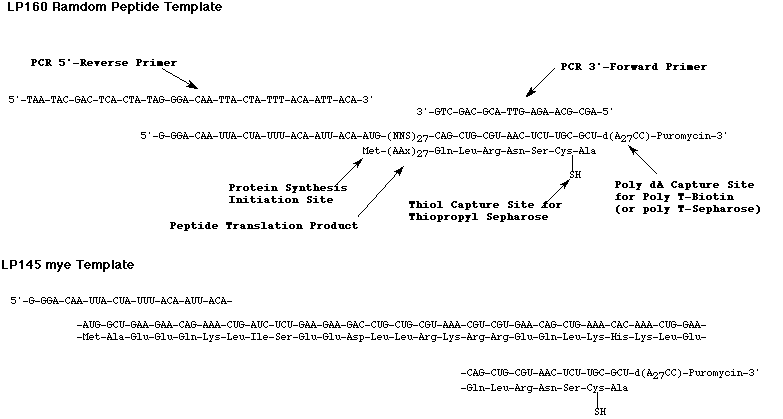
To demonstrate that the method could be used for selection enrichment, the authors synthesized two mRNA-puromycin peptide templates; LP160 random peptide template and LP154 myc template (see Figure). A number of common features were built into each template:
- A 5' sequence that promotes ribosome binding and translation initiation.
- An AUG codon to initiate polypeptide synthesis.
- A mRNA coding sequence for either a known (myc) or random peptide.
- A cysteine residue to introduce a thiol capture site, only seen in a successfully translated peptide, for capture by thiopropyl Sepharose.
- A poly dA capture site for oligo-dT-biotin.
- Forward and reverse priming sites for PCR amplification of the reverse transcribed DNA sequences.
The templates were then translated in a reticulocyte lysate system to yield the peptide-RNA fusion product which was then isolated by sequential purification using dT25-biotin or dT25-Sepharose followed by thiopropyl Sepharose. The RNA was reverse transcribed using Superscript reverse-transcriptase to give a RNA-DNA duplex. Peptides synthesized using the LP154 myc template were selected using an anti-myc antibody. The selectivity of this method was demonstrated by immunoprecipitation of myc-RNA fusion product from up to a 200-fold excess of LP160-fusion product. Following selection the RNA-DNA duplex was heated in the presence of ammonium hydroxide to degrade the RNA. After removal of ammonium hydroxide by evaporation, PCR primers were added and the DNA amplified by PCR for subsequent sequencing.
Conclusion
Highly complex libraries of random peptide sequences linked to their encoding RNAs can be easily synthesized. The RNA-peptide fusion products carry their structural identity in a form that can be easily recovered following an activity-specific selection protocol. The ability to synthesize very specific RNA sequences from chemically or enzymatically synthesized oligonucleotides along with the availability of puromycin-CPG open up this exciting technique to everyone.
References:
- S. Borman, C&EN, Feb. 12, 1996, 29-54.
- R.W. Roberts and J.W. Szostak, Proc. Natl. Acad. Sci. USA, 1997, 94, 12297-302.