Glen Report 22.19: Technical Brief - Depurination
Depurination (cleavage of the glycosidic bond connecting the purine base to the sugar) is a term that is not mentioned very much in the context of regular oligonucleotide synthesis - probably because the optimized processes used on modern synthesizers do not really induce depurination. Depurination is more likely to occur in the base protected monomers used for oligonucleotide synthesis than in the nucleosides themselves due to the electron withdrawing effect of acyl protecting groups which then destabilizes the glycosidic bonds. Ribonucleosides with their additional 2'-OH groups are less susceptible than deoxynucleosides. At Glen Research, we are concerned about depurination (and associated depyrimidination) when dealing with unusual modified nucleosides that are more susceptible to this degradative process.
The consequence of depurination during oligonucleotide synthesis is the loss of the purine base to form an internucleotide linkage containing the abasic sugar at that position. This site is stable during further synthesis cycles but, on deprotection with basic reagents, the oligonucleotide is cleaved at that position leading to two shorter fragments. The fragment towards the 5' terminus still contains the DMT group often used for DMT-ON purification. Following such purification, the depurinated fragments remain in the product as oligonucleotides truncated towards the 3' terminus. Clearly, this is not a desirable outcome.
Formamidine Protection
One key innovation in protecting groups for oligonucleotide synthesis was the formamidine (Figure 1) protecting group.1-3 Acyl protecting groups are electron withdrawing and destabilize the glycosidic bond. Formamidines are electron donating and stabilize the glycosidic bond. Formamidines first appeared in commercial DNA synthesis with the introduction4 of the dimethylformamidine (dmf) group in dmf-dG (and dmf-dA) as “Fastphoramidites”. The adoption of dmf-dG was immediate but dmf-dA proved to be rather too labile for routine use and was discontinued.
It is worthy of note that the formamidine family is more stabilizing with increasing length of the alkyl groups. At the same time, formamidine groups become more resistant to deprotection with increasing length of the alkyl groups. So dibutylformamidine (dbf) is significantly more stabilizing than dmf but dbf deprotects much slower than dmf. Using formamidine protecting groups, a variety of minor bases have become amenable to use in oligonucleotide synthesis. For example, the iso-bases, iso-dC and iso-dG, are both very susceptible to acidic cleavage and are only usable today because of formamidine protecting groups.
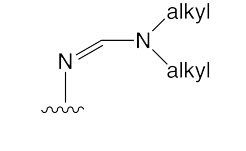
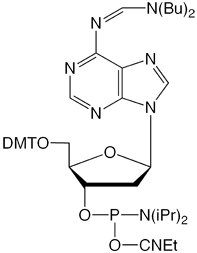
dbf-dA-CE Phosphoramidite (1)
Deblocking Acids
Many years ago, in an effort to mimic the conditions of synthesis of long oligos or those produced on a large scale, we conducted experiments where oligonucleotides growing on CPG were subjected to long wait periods in the presence of a deprotection solution containing trichloroacetic acid (TCA). We duly detected depurination at dA sites but not at a very high level. However, when we used a deprotection solution containing dichloroacetic acid (DCA), we were unable to detect any depurination. It was clear that in routine oligonucleotide synthesis, even of long oligos, on a regular column-based synthesizer, depurination is not a very significant event.
A recent report5 has reminded us that depurination may not be significant in normal synthesis but it is still a problem in certain circumstances. Indeed, LeProust and collaborators have shown that depurination is the limiting factor in the ability to synthesize long oligos (up to 150mers) on Agilent's DNA microarray synthesis platform – even using DCA as the detritylating reagent. The authors note that the difference between a flow-through synthesis on the porous, 3-dimensional surface of CPG (where the support surface area is large and the reagent volume is small) and the synthesis on the flat, non-porous surface of a silicon wafer (where the surface area is low and surrounded by a high volume reagent chamber) is profound. In this case, it was shown that the detritylation conditions were not a significant factor in the level of depurination, which was a little unexpected. Depurination had to be controlled by adjusting the fluidics of the flow cell and by quenching the detritylation solution with oxidizer solution.
For researchers trying to determine if their source of depurination is reagent, fluidics or protocol-based, we are happy to turn the clock back more than two decades and offer a depurination-resistant dA monomer. Based on projected pricing, this is unlikely ever to replace the regular Bz-dA monomer but we hope that dibutylformamidine-protected dA (dbf-dA-CE Phosphoramidite) (1) will prove useful in this basic research. Dbf-dA is compatible with UltraFast deprotection and is deprotected by ammonium hydroxide at 55 °C overnight.
References
- B.C. Froehler, and M.D. Matteucci, Nucleic Acids Res, 1983, 11, 8031-6.
- L.J. McBride, R. Kierzek, S.L. Beaucage, and M.H. Caruthers, J. Amer. Chem. Soc., 1986, 108, 2040.
- Z. Tocik, L. Arnold, and J. Smrt, Nucleic Acids Res., 1987, 18, 193.
- H. Vu, et al., Tetrahedron Lett., 1990, 31, 7269-7272.
- E.M. LeProust, et al., Nucleic Acids Res., 2010, In Press.
Product Information
dbf-dA-CE Phosphoramidite has been discontinued.