Glen Report 19.12: 1-Methyl-Adenine in Nucleic Acids
Natural nucleic acids, in particular RNA, contain a vast quantity of modified nucleosides1 that are responsible for a large variety of chemical and potential biological functions. Some of these modifications are biologically essential, while some others are detrimental and mainly considered as lesions. In this article, we present new additions to our product portfolio and describe how similar base modifications may have completely different consequences when in RNA or DNA.
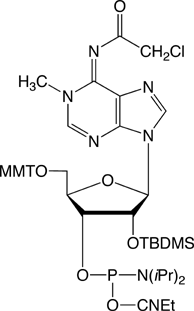
1-Methyladenosine
Even though nucleic acid modifications have been linked to control of gene expression at both the levels of transcription and translation through folding and alternate foldings,2,3 much has still to be done to understand the contribution of modified nucleosides to the functional chemistry, structure and biological activity of RNA. Consequently, Glen Research continues to evaluate the potential of novel nucleoside phosphoramidites to help this research to progress. We are particularly happy to introduce here 1-Methyladenosine (m1A) as a phosphoramidite for RNA modification.4
Methylation of adenosine at position 1 produces a drastic functional change in the nucleobase. 1-Methyladenosine (pKa 8.25) is a much stronger base than adenosine (pKa 3.5). N-1 methylation excludes participation of the adenine base in canonical Watson–Crick base pairing and provides a positive charge to the nucleobase. This modification also alters the hydrophobicity of the base, the stacking properties, the ordering of water molecules and the chelation properties. The base may become involved in non-canonical hydrogen bonding, in electrostatic interactions and, in general, it may contribute to the conformational dynamics of the tRNA.
1-Methyladenosine (m1A) is obtained in nature by post-transcriptional methylation of adenosine by methyl-1-adenosine transferase5 and it has a special role in t-RNA folding.6,7 Approximately 25% of all tRNAs have m1A at position 58 in the T loop, while m1A also often occurs at position 14 in the D loop.8
Reverse transcription is a central step in HIV-1 replication that represents a typical case of interplay between viral and cellular factors. HIV-1 diverts a cellular tRNA, tRNALys3, to prime reverse transcription. The post-transcriptional modifications of tRNALys3 are crucial for completion of reverse transcription. In all HIV strains, methylation of A58 is required to allow productive strand transfer during (+) strand DNA synthesis.9
The function of modified nucleosides is usually studied by comparison of the properties of modified tRNAs (purified from wild-type organisms) with that of unmodified tRNAs (obtained by in?vitro transcription) and of partially modified tRNAs (from modification mutants).
Because m1A is a basic nucleoside, which can easily undergo a Dimroth rearrangement to yield N6-methyladenosine, the design of a system for its incorporation into synthetic oligonucleotides is not easy. Here we are happy to propose a protocol first described by Sergey Mikhailov, Piet Herdewijn and coworkers4 from the Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow (Russia) and the Laboratory of Medicinal Chemistry, Rega Institute, Katholieke Universiteit Leuven (Belgium). These groups described the synthesis of the phosphoramidite of protected 1-methyladenosine (1) and its successful incorporation into RNA. The availability of this m1A phosphoramidite will now enable researchers to synthesize substrate or prototype more easily.
This new RNA minor bases is a 2‘-TBDMS protected phosphoramidite. Synthesis tests have been performed on ABI 39x synthesizers and using either standard (N-Bz-A/N-iBu-G/N-Ac-C) or the mild deprotection phosphoramidites (N-Pac-A/N-iPr-Pac-G). We tested several coupling times and several activators and found that 0.25M 5-Benzylthio-1H-tetrazole in acetonitrile gave the best coupling efficiency. With this activator we obtained 96% coupling with a coupling time of 15 minutes, while we barely obtained over 90% coupling after 15 minutes with 1H-tetrazole.
Deprotection of the bases is then carried out in 2M methanolic ammonia at room temperature for 17 - 48 hours depending on the base composition of the oligo. Mikhailov and co-authors4 found that 60 hours at RT is sufficient to cleave the N2-isobutyryl protecting group of the guanine base. However, we prefer the UltraMild approach in order to avoid any degradation of the fragile m1A.10 Also, we would recommend the use of the UltraMild Cap A reagent containing phenoxyacetic anhydride (Pac2O). This modification removes the possibility of exchange of the iPr-Pac protecting group on the dG with acetate from the regular acetic anhydride capping mix. After deprotection, the solution was decanted from the support, the methanolic ammonia was evaporated, and the residue was treated with 1.5 mL of a 1M TBAF solution for 18 hours at room temperature.
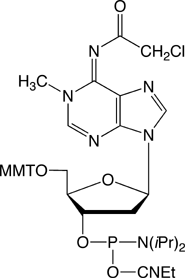
1-Methyl-2’-deoxyadenosine
As described above, alkylation may be desirable or may be a problem. Alkylating agents are abundant in the environment and are also generated inside cells. Such agents introduce various and numerous lesions in DNA and some are even carcinogenic in mammals. To counteract the effects of these lesions, most organisms express several mechanisms for repairing alkylation damage in DNA.
In brief, cellular polynucleotides are alkylated by endogenous components, such as S-adenosylmethionine, or after reacting with two general classes of environmental and laboratory chemicals. SN1 agents include alkylnitrosourea (e.g., MNU, ENU) and N-alkyl-N-nitro-N-nitrosoguanidine (e.g., MNNG) that react with the N7 position of guanine, N3 of adenine, O6 of guanine, O2 or O4 of pyrimidines, and the non-phosphodiester oxygen atoms of the phosphate backbone. In contrast, SN2 agents such as methyl methanesulfonate and dimethyl sulfate react primarily with the N1 position of adenine (1-Methyl-2’-deoxyadenosine) and N3 of cytosine.11
To overcome the mutagenic and toxic effects of these modifications, cells produce a variety of DNA repair enzymes. For example, E.coli ogt and ada genes encode O6-methylguanine methyltransferases, while ada also encodes O4-methylthymine methyltransferase and methylphosphotriester methyltransferase activities, and alkB encodes an oxidative demethylase that directly reverses 1-alkyladenine and 3-alkylcytosine lesions in DNA or RNA.12,13
The availability of m1A 2‘-deoxy-nucleoside (m1dA) phosphoramidite (2) will now enable researchers to synthesize substrate or prototype more easily.
The m1dA phosphoramidite coupled very well (>99%) at all coupling times tested with 0.45M 1H-Tetrazole in acetonitrile as activator, so a standard coupling method can be used for this amidite. We conducted a series of deprotection experiments on the deoxynucleoside to confirm the literature results and to see if we could use an alternate deprotection reagent. Since this m1dA is sensitive to rearrangement to m6dA when using standard Nh4OH deprotection methods, we recommend the use of UltraMild phosphoramidites for the synthesis of oligonucleotides containing this sensitive modified base. We have found that deprotection of oligos containing m1dA using 2M anhydrous ammonia in methanol gave good results and a 24h deprotection time seems to be optimal. Again, the use of the UltraMild Cap A containing phenoxyacetic anhydride (Pac2O) is also to be recommended.
Other DNA Repair Substrate
This addition to our catalogue is just another example of our commitment to provide the researcher interested in DNA damage and repair with the largest range of products in this field. We would like to review these products as follows.
As mentioned above, DNA exposed to reagents like nitrosourea or dimethylsulfate result in different sorts of alkylation lesions. Most of these lesions modify the base pairing properties of the nucleobase (e.g., DNA polymerases readily insert T opposite O6-methyl-dG). We offer amidites that enable the incorporation of O6-methylguanine, N-6 methyladenine and O4-methylthymine.
The 8-oxo purine monomers allow investigation of the structure and activity of oligonucleotides containing an 8-oxo mutation, which is formed naturally when DNA is subjected to oxidative conditions or ionizing radiation.
5,6-Dihydro pyrimidines are naturally occurring compounds that are structural components of alanine transfer RNA. Dihydrouracil and the hydroxy pyrimidines are major base damage products formed by exposure of DNA to ionizing radiation.
8-Amino-G is formed along with 8-oxo-G as the major mutagenic lesions formed in DNA damage caused by 2-nitropropane. 2-Nitropropane is an industrial solvent and a component of paints, dyes and varnishes, and is also present in cigarette smoke.
Thymine glycol (5,6-dihydroxy-5,6-dihydrothymine) is formed when thymine is subjected to oxidative stress, including ionizing radiation. Oxidation of the 5,6 double bond of Thymidine generates two chiral centers at C5 and C6. The cis-5R,6S form is generated as the predominant product along with the other diastereomer, the cis-5S,6R form. The presence of thymidine glycol in DNA has significant biological consequences and many organisms possess specific repair enzymes for the excision of this lesion.
2-Aminoimidazolone (Iz) and its hydrolysis product imidazolone (Z) are major oxidation products of G. Access to these two potential lesions is not possible during oligonucleotide synthesis because they are so base-labile. A suitable precursor, 8-methoxy-dG (8-OMe-dG), to dIz has now been described. The conversion of 8-OMe-dG to dIz takes place by irradiation of the oligonucleotide (1 mM) in 50 mM sodium cacodylate buffer, pH 7, in the presence of riboflavin (50 µM) for 2 minutes on a transilluminator (366 nm), under aerobic conditions at 4°C. Interestingly for a photochemical reaction, the conversion is virtually quantitative.
Hydrolysis of nucleoside residues in DNA occurs naturally to generate abasic sites. Most commonly, dA sites are hydrolyzed causing depurination and leading to abasic residues. A new chemical method allows the generation of abasic sites in double and single stranded oligonucleotides using very mild specific conditions and with very low probability of side reactions. The abasic phosphoramidite allows oligonucleotide synthesis under standard conditions. Following standard deprotection, the silyl protecting groups of the residue are removed with aqueous acid. (This can be done in conjunction with trityl removal in the last step of a DMT-on purification.) The diol so formed is then treated with aqueous sodium periodate to form an aldehyde plus formaldehyde. The aldehyde then immediately cyclizes to its preferred structure, the abasic cyclic sugar (dR). dSpacer has also been used successfully as a mimic of the highly base-labile abasic site.
Finally, one of the major sources of DNA damage in all organisms is the UV component of sunlight. The predominant reaction induced by UV light on DNA is dimerization of adjacent pyrimidine bases leading to cyclobutane dimers (CBDs). The dimers formed in the most significant quantity are the cis-syn cyclobutane dimer of two thymine bases. Although formed routinely, these dimer products are efficiently excised and repaired enzymatically (nucleotide excision repair) or the dimerization is reversed by photolase enzymes. These lesions have been connected to the formation of squamous cell carcinomas. In addition, humans who lack ability to repair CBD lesions with high efficiency may be genetically predisposed to Xeroderma Pigmentosa (XP), a disease characterized by extreme sensitivity to sunlight and high frequency of skin cancer. Polymerases encountering unrepaired CBD lesions are quite error-prone, leading to incorrect base insertions and subsequent mutations.
References
- J. Rozenski, P.F. Crain, and J.A. McCloskey, Nucleic Acids Res., 1999, 27, 196-197.
- P.F. Agris, Prog Nucleic Acid Res Mol Biol, 1996, 53, 79-129.
- M. Helm, Nucleic Acids Res., 2006, 34, 721-733.
- S.N. Mikhailov, et al., Nucleic Acid Res., 2002, 30, 1124-1131.
- J. Anderson and L. Droogmans, Biosynthesis and function of 1-MeAdo in tRNA. In Topics in Current Genetics, Springer Eds: 2005; 12, pp 121-139.
- M. Helm, et al., Nucleic Acids Res., 1998, 26, 1636-1643.
- R. Sengupta, et al., Nucleic Acids Res., 2000, 28, 1374-1380.
- H. Sierzputowska-Gracz, H.D. Gopal, and P.F. Agris, Nucleic Acids Res., 1986, 14, 7783-7801.
- R. Marquet and F. Dardel, Transfer RNA modifications and DNA editing in HIV-1 reverse transcription. In Topics in Current Genetics, Springer Eds.: 2005; 12, pp 401-429.
- E.N. Timofeev, Sergey N. Mikhailov, Andrei N. Zueva, Ekaterina V. Efimtseva, Piet Herdewijn, Robert L. Somers and Marc M. Lemaitre, Helv Chim Acta, 2007, 90. in press.
- B. Sedgwick and T. Lindahl, Oncogene, 2002, 21, 8886-94.
- S.C. Trewick, et al., Nature, 2002, 419, 174-8.
- P. Falnes, R.F. Johansen, and E. Seeberg, Nature, 2002, 419, 178-82.
Product Information
1-Me-A-CE Phosphoramidite (10-3501)
1-Me-dA-CE Phosphoramidite (10-1501)