Glen Report 13.13: Trityl Group in the 3rd Millenium: New Perspectives for Oligonucleotide Chemistry and Beyond
M.S. Shchepinov, Sequenom, Inc., Email: misha@bioch.ox.ac.uk
Abstract (or what can now be done with trityl-based compounds)
Fluorescence
- Novel trityl-based fluorescent labels incorporating polycyclic aromatic hydrocarbons (PAHs), such as pyrene and perylene, allow controlled activation/deactivation of fluorescence (Section 2.1).
- Trityl-based labels are useful tools for surface chemistry applications, such as oligonucleotide array/DNA chip technology (Section 2.1).
- The use of these tritylized fluorescent tags in fluorescence resonance energy transfer (FRET) allows an additional degree of control of the energy transfer process, which could be beneficial in studying phenomena like DNA charge transfer and electroluminescence (Section 2.2).
Combinatorial chemistry and mass-spectrometry
- Extremely high desorption properties of trityl-based compounds in mass-spectrometry, partially due to the high stability of the cations, make them valuable as encoding tags for combinatorial synthesis (Section 3.1).
- Mass-tags forming a monolayer on the surface can still be unambiguously detected by mass-spectrometry, suggesting numerous applications in surface sciences, including oligonucleotide array/DNA chip technology (Section 3.1).
- Unique desorption efficiency of trityl mass-tags even in a mixture makes them extremely useful for calibrating mass-spectrometers with a very high degree of precision, consistent with modern demands for 1-5 ppm range measurements (Section 3.2).
1. Introduction
That the trityl group is full of surprises was first demonstrated one hundred years ago this year, when it became the first stable free radical to be discovered1. The positive charge on the α–carbon atom is stabilized by the resonance effect of three aromatic rings (Scheme 1A). This particular property, which makes trityl ethers acid-labile, turned out to be useful, and the next few decades saw the trityl being developed into a major class of protective groups widely used in nucleoside2, oligonucleotide3, peptide, carbohydrate, and indeed in almost all other fields of organic and bioorganic chemistry4. Trityl-based compounds also occupy an important niche in organic dye chemistry. More recent applications in the life sciences include multi-color monitoring of oligonucleotide synthesis yields5 and the use of a modified trityl group as a cleavable cross-linking moiety, for example, to purify oligos by immobilizing on to a solid support after synthesis either through a Diels-Alder6 process or an amide bond formation7. Modified trityl groups have also been used to accelerate the formation of internucleotide bonds in the phosphotriester approach8, to quantify the amount of amino groups on a solid support9, and to controllably activate pro-drug antibody conjugates10. A modified trityl group bearing a pyrenyl residue in place of one of the aryl groups has been used for more precise fluorescent detection (down to 10-10 M) of detritylation11, and a 14C-labelled 4,4'-dimethoxytrityl (DMTr) group was used for more sensitive monitoring of coupling reactions on an aminated polypropylene support12. Some new derivatives of the trityl group and their applications are discussed below.
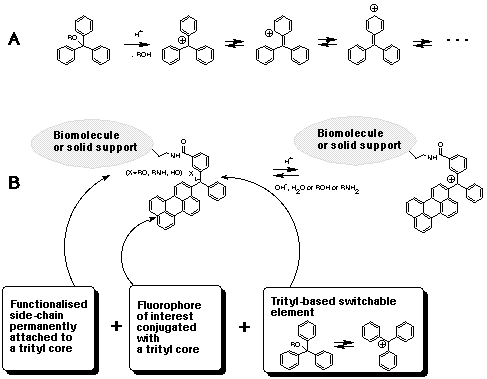
2. Fluorescence
2.1 Tritylization of Polycyclic Aromatic Hydrocarbons (PAHs): a New Family of Switchable Fluorescent Labels
Both single fluorophore13 and energy transfer14 based fluorescence detection methods find wide applications in the analysis of nucleic acids. Some PAHs have certain advantages over the fluorophores, such as fluorescein, currently used to label biomolecules: they are less prone to photobleaching and have high molar absorbance and quantum yields. They are also chemically more stable and do not degrade in the conditions of oligonucleotide and peptide synthesis. Furthermore, molecules are available with a range of excitation and emission maxima and large Stokes shifts. Another potentially useful feature is sensitivity to chemical environment. This is especially so for pyrene and perylene, which make them PAHs with very useful fluorescent properties. The introduction of a trityl-based carbinol-carbocation switchable element to these PAHs would allow their fluorescence to be controllably turned on and off by changing pH. In addition, the conversion of these PAHs into trityl-type structures would be advantageous due to the non-planar conformation of triarylmethanols (the angle of twist is ca 35° for a cation15 and is even bigger for tritanol), which would prevent the p-p stacking interactions with a resulting increase in the solubility.
A modified trityl group bearing a pyrenyl residue in place of one of the phenyls has fluorescent properties similar to non-modified pyrene11. The cation derived from the triarylmethyl group on acid treatment has substantially red-shifted spectral characteristics due to the conjugation of all aromatic rings through the cationic central α–atom. The cation will still remain covalently linked to a probe molecule if attached to it through a side-chain, as shown in Scheme 1B16. These features are combined in compounds 1 and 2 (Scheme 2), which are designed to label amino group(s).
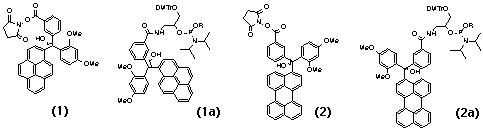
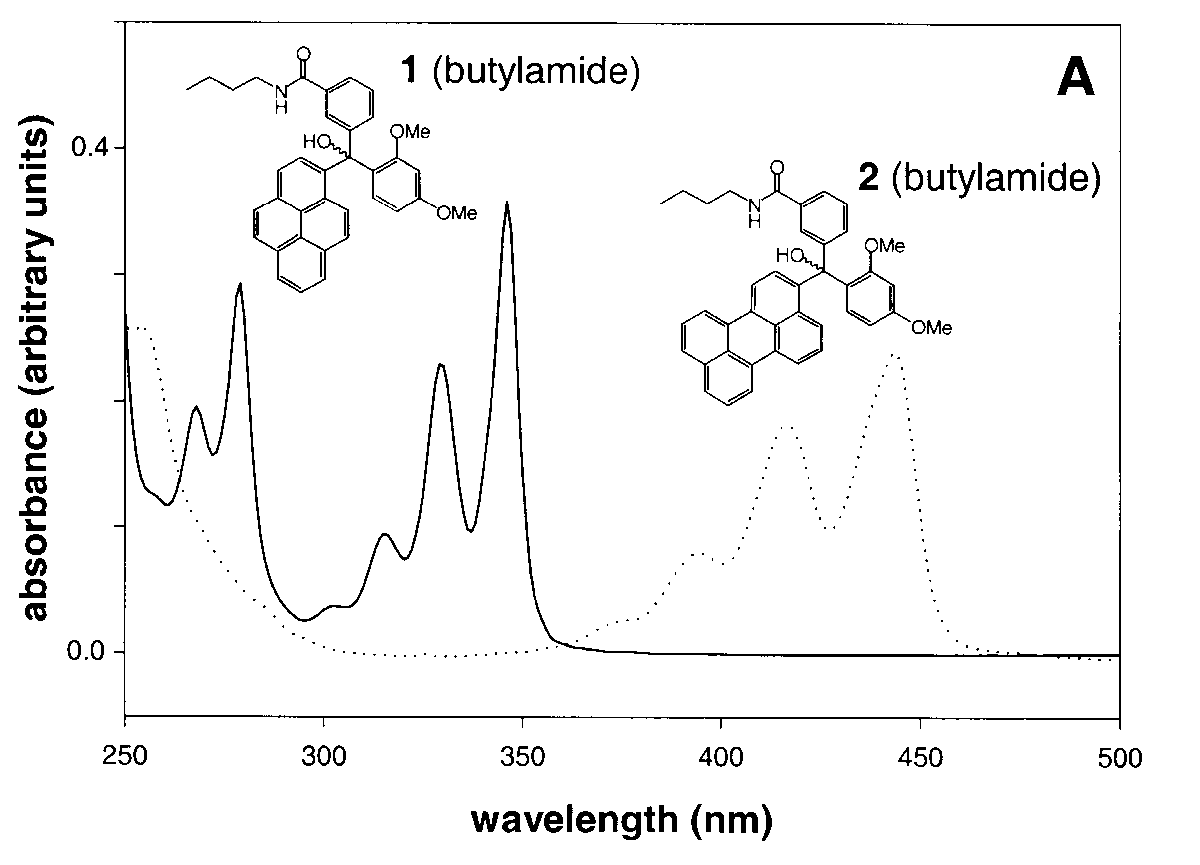
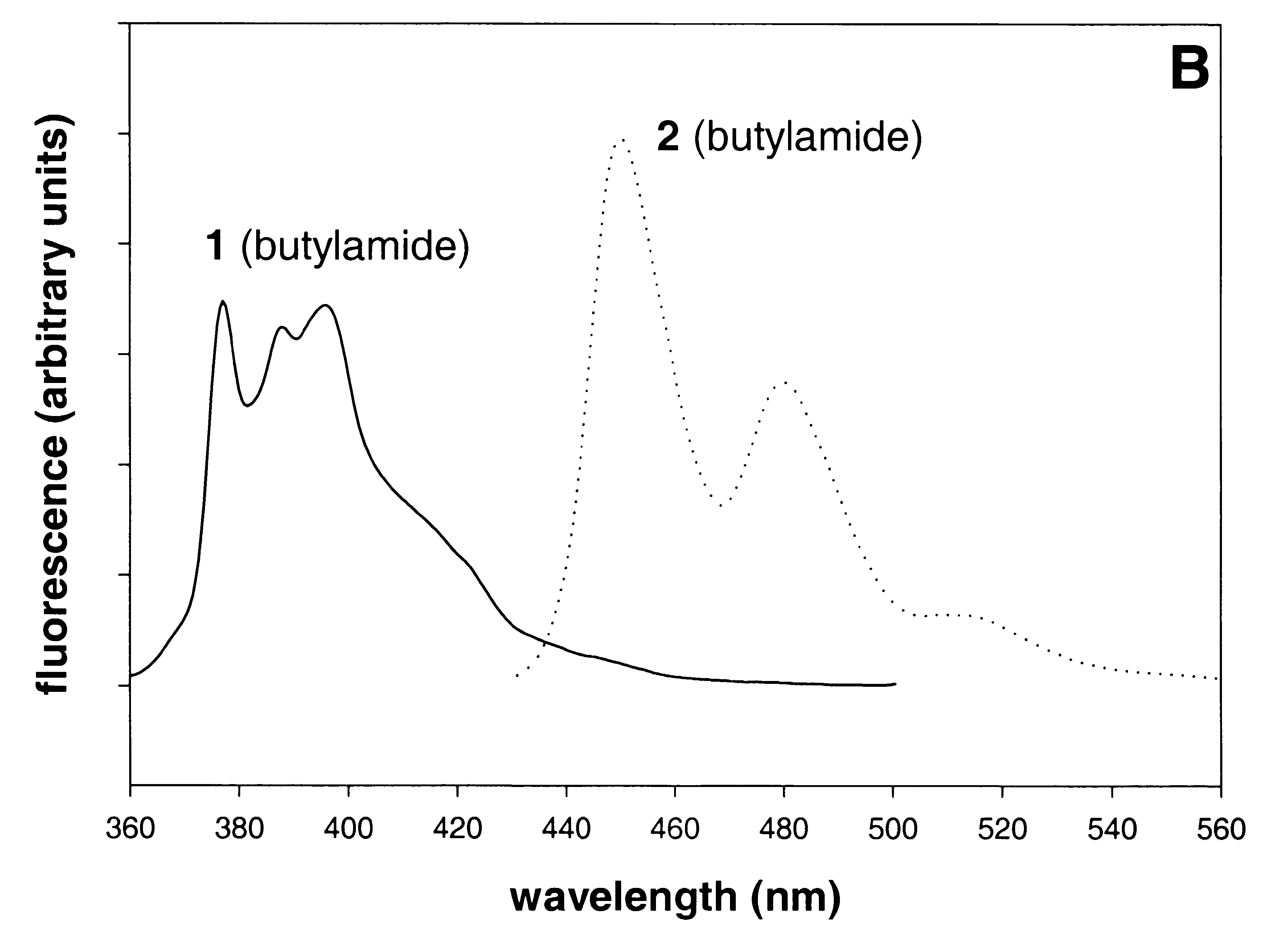
The UV spectra of model (butyl) amides based on 1 and 2 (Figure 1A) are the same shape but slightly red-shifted (3-10 nm) relative to the starting PAHs (Figure 1A). Their fluorescence spectra are shown in Figure 1B. The pH-threshold for the formation of trityl carbocations (Scheme 1) from corresponding tritanols at low pH can be controlled by electron withdrawing or donating groups in the aromatic rings3. Two methoxy groups and one carboxyl group give 1 and 2 an acidic stability similar to that of the 4-monomethoxytrityl (MMTr) group.
Compounds like 1 and 2 (Figure 1A and 1B) can be used to expand the palette of fluorophores for multicolor DNA detection on DNA chips. Multicolor detection (use of more than one fluorophore in one reaction) is a useful feature of fluorescent dyes; it enables different sequences to be detected simultaneously and was employed in DNA sequencing13, FISH17, and gene expression analysis on DNA chips18. The size of the palette is limited by the overlap of the excitation and emission spectra and it has proved difficult to use more than four colors in FISH, two colors being more normal in expression analysis. An advantage of trityl-based fluorescent tags is the potential to 'switch' the spectra on and off by simply changing the pH. The magnitude of the shifts is very large. For example, moving from neutral or alkaline to acidic pH shifts the excitation maximum of the pyrene-based compound 1 from 346 to 711 nm (Figure 1C).
Trityl carbocations do not fluoresce in the range detected for the corresponding tritanols. This property can be used to improve the discrimination of labels: first by increasing the accuracy of intensity measurements; and, second, increasing the potential number of colors in the palette. For example, targets can be labelled with two fluorophores having similar excitation and emission spectra, but only one of which is switchable by pH change. After hybridization, measurements are taken at two pH values: under ambient conditions, and after exposing the array to acidic vapor, which is enough to switch the emission of fluorescent trityls off immediately but reversibly. Using a single excitation source, both fluorophores emit at neutral pH but only one will emit in acid. These two measurements alone would be enough to distinguish the two patterns of hybridization. But a third measurement, using a source which excites the second fluorophore in acid, allows even more analysis. In this way it may be possible to double the number of labels that can be used together.
Cyanoethyl phosphoramidites 1a and 2a (Scheme 2, R=CNEt) are suitable for standard oligonucleotide synthesis and deprotection. However, if it is desirable to have the labels attached through a non-charged linker, then the ethyl phosphoramidites (R=Et) should be employed, since they can yield non-charged phosphotriesters after ammonolysis.

2.2 Energy Transfer Applications: No Fret with FRET
To evaluate the suitability of compounds 1 and 2 as components for FRET, a model compound 3 (Figure 2A) was prepared. While the absorption spectra for both non-ionized and bis-cationic forms (Figure 2A) essentially represent a superimposition of 1 and 2, compound 3 fluoresces only at 450, 480 and 515 nm when excited at the pyrene absorption maximum of 330 nm (Figure 2B), with no detectable fluorescence of pyrene (at 377, 388 or 396 nm). When mixed in equimolar amounts, model amides of 1 and 2 (Scheme 2) retain their own fluorescence properties. This suggests a possibility of designing fluorescent labels having increased Stokes shifts by arranging the necessary fluorophores (perhaps even more than two) in the vicinity of each other19. Furthermore, an additional control can be achieved by making some of these parts more acid-labile than the others, so that some selected components of the chain may be reversibly switched off by decreasing the pH.
Interestingly, MALDI-TOF MS of 3 shows fragments lacking one (1193.46) and two (1176.47) hydroxyl groups. The fact that the latter flies in mono-charged (mono-cationic) and not in bis-cationic form (the signal for 1176.49/2 = 588.25 was not detected) suggests some unusual interactions, perhaps FRET during the LDI-TOF process, initiated by laser irradiation at 340 nm, which is almost a perfect match with the absorption maximum for 3.
Applications may be possible for such trityl tags (probably in more acid-labile form, with three or more methoxyls) in the burgeoning field of DNA charge transport20, where methods are needed for monitoring carrier mobility (or lack of it) through the stacked aromatic 'core' of a double helix.
The fluorophore-containing trityls attached to solid surfaces, such as glass, silicon oxide, polypropylene, can be used to monitor local environmental changes, such as pH, as previously described16. The ability of a trityl group to ionize under the influence of light25 can be utilized to reversibly produce monolayers of these species on the surfaces. The field of electroluminescence21 can also benefit from using the compounds which combine the hole-transporting properties (trityl) with the presence of moieties routinely used as dopants for polyphenylenevinylene (PPV) (perylene for blue color) and are easy to attach (side-chain).


3. Applications in Mass-Spectrometry
3.1 Trityl Mass-Tags: Encoding of Combinatorial Libraries27
The combinatorial approach to the simultaneous synthesis of large numbers of compounds on solid supports has been an important recent development in biological and medicinal chemistry22. Two methods predominate: spatially addressable arrays, in which synthesis steps are performed simultaneously on sets of physically separated starting materials or areas23; and bead libraries, consisting of mixtures of microscopic resin beads each of which carries a single compound, usually synthesized using the 'split-and-mix' method24. Although resin libraries are quickly screened, their application is limited if the compound on a selected 'hit' bead cannot be readily identified.
One way around this is to attach one or more tags to the bead, which can be cleaved and identified even at very low concentration, and which will encode the synthesis steps that the bead has undergone. An encoding tag must survive the synthesis and assay, be cleaved specifically and orthogonally from the tethered compound, and be readily identifiable in pmol or fmol quantities. The problem of encoding during 'one-bead-one-compound' combinatorial synthesis has been addressed by several groups (the methods of identification of the tags include HPLC, GC, MS, IR, and NMR) and recently reviewed22. However, none of these methods has been particularly successful for encoding large libraries, such as those formed by a complete set of all possible oligonucleotides of a defined length. The sheer numbers involved point to the use of bead libraries for their synthesis and screening, but the limited sensitivity of gel-based sequencing methods rules out direct identification.
The characteristic signal of the DMTr cation (monoisotopic peak, exact mass 303.139 Da) is frequently present in mass-spectra of DMTr-containing compounds, suggesting that derivatives of trityl groups with different masses could serve as unique markers in combinatorial synthesis27. This unique application is based on the high desorption rate of trityl cation-based tags in the conditions of LDI-TOF MS, which makes detection simpler than in previously described encoding systems22. Trityl cations can be released by acidic treatment and detected by LDI-TOF analysis with or without a matrix. Alternatively, the cations can be generated directly by laser irradiation25, which permits direct detection of tagged DNA on surfaces, for example, when hybridized at different positions of a DNA chip.
Trityl-based synthons 4 and 5 (Scheme 3) are suitable for use as mass-markers. To produce the mass-tags 4a and 5a, these synthons are treated with different amines.
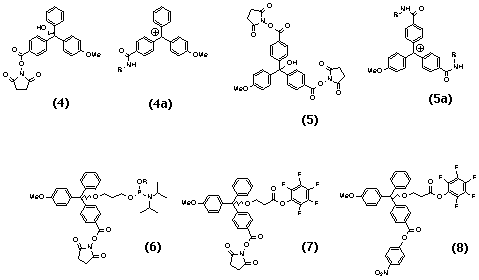
The masses of the majority of cheap commercially available primary amines which would withstand the conditions of oligonucleotide synthesis and deprotection (thus excluding, for example, all aromatic amines unless 'fast' phosphoramidites are used for the combinatorial synthesis) lie mainly in the range of 50-300 Da. For some applications it is desirable to have several hundred mass-tags available. The resolution of the tags in TOF mass-spectrometry is satisfactory with �2 Da difference between the masses of tags. Secondary amines are not useful because they are less reactive and usually there are primary amines with the same masses. Amines bearing other reactive groups cannot be used as tags in the conditions of oligonucleotide synthesis unless they are introduced at the very last step or an additional capping step is employed. Therefore, the above range of amines can only yield a limited number of tags.
The compound 5 allows attachment of two amines to the same trityl moiety, thus extending the series of mass-tags into the higher mass range (Scheme 3). NHS-activated trityls react with amines in THF or dioxane to give well-flying mass-tags. A typical LDI-TOF spectrum is shown in Figure 3. The acidic stability of the corresponding ethers is as follows: MMTrOR < MMTr(NHS)OR < MMTr(2NHS)OR ~ Tr and the MMTr(NHS) group is more than 50 times more stable to acid than the DMTr group. However, no difference was detected in the signal intensity of tritanols as compared to the corresponding trityl ethers when using laser ionization instead of acidic treatment, suggesting photocleavage by the laser irradiation25 as a good alternative to acidic cleavage.
Dilution experiments showed that the lower limit of (MA)LDI-TOF (either with or without matrix) detection of trityl-based tags is around 10-13 M concentration level. The diameter of the spot on the sample well that is covered by the laser beam is about 100-300 micron, which means that the actual amount of sample analyzed is in the fmol range. About 5% of sites are occupied by tags (Scheme 4), which is more than enough for detection.

To be used as a tag in oligonucleotide synthesis, the trityl group should give clean, high-intensity signal in (MA)LDI-TOF analysis. It should also survive several steps of the acidic treatment used to remove the 5'-DMTr group in oligonucleotide synthesis, that is, be orthogonal to the groups involved. The MMTr(NHS) group 4 (Scheme 3) remains attached to a primary hydroxyl group after at least 8-9 cycles of acidic deprotection in oligo synthesis under conditions described below, and is easily released as 4a using 1-3% TFA.
To introduce a tagging moiety during oligonucleotide synthesis (Scheme 4), the phosphoramidite synthon 6 (Scheme 3) was prepared. Based on a propanediol structure, compound 6 provides reactivity similar to that of standard 2'-deoxynucleoside phosphoramidites. The molar ratio between 6 and a standard amidite, with which it is premixed in solution, will therefore be maintained on the solid support after condensation. The phosphoramidite 6 is stable in acetonitrile solution at room temperature for at least 2 days.
Standard A, C, G and T phosphoramidites (either 3' or 5') are premixed with ca 3-6 mol% of 6 prior to oligonucleotide synthesis. Assuming the stepwise yield of oligonucleotide synthesis to be about 99%, for an 8-mer library synthesized using 6 as a 5% additive to all bases, ca 60% of all sites of the beads are occupied by full length oligonucleotides in the final product. The concentration of the first tag (5% of all initial sites) would be about two-fold greater than that of the last tag (5% of the remaining 60% of the sites), which still makes it possible to detect all of them in the same mixture. Split-and-mix synthesis of oligonucleotides can be carried out in an Applied Biosystems 394 DNA/RNA four column synthesizer. The solid support used was Tenta Gel Macrobeads OH, 280-320 mm, polystyrene beads grafted with polyethyleneglycol chains (Rapp Polymere), but other supports can also be employed. The natural loading of the beads can be dramatically increased by employing the Trebler phosphoramidite26 (Glen Research), each addition of which triples the amount of reactive groups on the beads. The beads were placed in four reusable polypropylene DNA synthesis columns (Glen Research). The oligonucleotide synthesis was carried out on a 1 mmol scale but the supply of deblocking reagent (diluted to 50% of its original concentration with CH2Cl2) to the columns was reduced to 10-15 sec, with a subsequent waiting step (10 sec). Thorough CH3CN washing ensured that all DMTr+ is desorbed. Before each detritylation step, the columns were washed with CH3CN in Manual Control mode, and then treated with corresponding amines (0.5 ml of 0.5-1 M solutions in dioxan/THF) for 1 min using 1 ml syringes, again washed with CH3CN and dried in vacuo for 15 min. The beads from all columns were then combined, mixed, and split again. The procedure was repeated until the end of the synthesis. The beads were then deprotected in concentrated ammonia and washed with distilled water.
At the end of the synthesis, each bead would contain a unique oligonucleotide sequence covalently attached to it. Beads can be selected by hybridization with labelled (for example, Cy5-labelled) oligonucleotide. After washing in the same buffer, the beads are transferred on to the surface of a microscope slide and the excess of the buffer removed by blotting with tissue paper. Colored (or otherwise identified beads) are then removed, washed with water at elevated temperatures, acetone and dried. The trityl tags can be cleaved with a few mL of 1-2% solution of trifluoroacetic acid in standard Deblock Solution (Glen Research; TCA/CH2Cl2) for 3-4 min. The supernatant is evaporated several times with acetone and methanol and the residue analyzed by (MA)LDI-TOF HRMS. The size of Rapp-beads (~0.3 mm) allows for manual removal of positively identified beads from the pool. For smaller beads, automated methods such as FACS might be used. An example of a decoding spectrum is shown in Figure 3.

To eliminate the problem of gradual loss of encoding MMTr-based tags 4 or 8 during the detritylation step in oligonucleotide synthesis, the use of Fmoc as a 5'-protecting group,28 (thus omitting an acidic treatment altogether) was also investigated. After each oxidation step, the columns were removed from the synthesizer, and the beads were treated with the corresponding amines, washed with CH3CN and then treated with 0.1 M DBU in CH3CN to remove Fmoc-protection. The tags encoding for up to 9-mer oligos synthesized using this strategy were detected using (MA)LDI-TOF analysis. Any other method employing non-acidically removed 5'-protective groups could also be used. For longer sequences, the 3'-ethyl or 3'-methyl phosphoramidites of 5'-Fmoc- (or other) protected nucleosides should preferably be used instead of cyanoethyl phosphoramidites, to prevent the loss of the CNEt protecting group due to the treatment with amines and DBU. Similarly, for longer sequences it is better to use the methyl derivative of 6 (Scheme 3).
The trityl mass-tags can also be used for encoding in combinatorial peptide synthesis. For that, the reagents 7 and 8 (Scheme 3) can be used, which have carboxyl groups activated to different extents: first, the more active pfp-activated group reacts with the amino-groups of amino-acids on the solid support in a way similar to that depicted on Scheme 4. The excess of the tagging amine then converts less reactive NHS-activated or pNP-activated carboxyl group into an amide, thus completing the encoding procedure.
3.2 Trityl Mass-Tags as 'Trityl Ladders': Calibration of Mass-Spectrometers and High Precision Mass Measurements
It has become standard practice in modern organic chemistry to characterize new compounds by mass-spectrometry with a precision of at least 5 parts per million. For a compound with the mass of a few hundred Da, this would mean a complete match between the theoretically calculated exact mass and that found for at least two digits after a decimal point. To achieve that degree of precision, one needs mass-markers which possess a very high desorption rate (in other words, fly well), and can potentially cover a long range of masses. Peptide-based mass-markers or the dextran derivatives presently used possess neither of these two properties to the desired extent.
Trityl mass-markers are easy to design and make just by treating activated trityl blocks like Tr(NHS) (9), MMTr(NHS) (4), DMTr(NHS) (10) and MMTr(2NHS) (5) with appropriate amines (Scheme 5). The pool of amines is not limited by the demands of stability to the conditions of oligonucleotide synthesis and deprotection, as is the case for the mass-tags, so aromatic amines and amines with functional groups can also be used. The exact masses for compounds with molecular weights of 350-800 Da were routinely measured with a precision of 0.5-1 ppm using trityl mass-tags as markers, whereas with standard peptides it was usually 5-7 ppm. For heavier samples, where the availability of amines becomes a limiting factor, the methods of synthesizing these amines can be developed based on solid-support synthesis.
The calibration curve for the majority of modern mass-spectrometers is not linear. It is therefore highly desirable to have more than two mass-markers in the same experiment. At the same time, the markers should not interfere with the analyte by decreasing its desorption rate. Mixtures of trityl-based tags ('trityl ladders') seem to be an ideal choice, since they possess an almost uniform desorption rate regardless of the amine used and can be detected at a really low concentration (Figure 4). With these properties, it looks like trityl-based mass-tags have a potential to become a mass-spec equivalent of the DNA/RNA ladders routinely used in gel electrophoresis!
Another potential application of the activated trityl blocks can be to activate compounds which are usually difficult to analyze, like carbohydrates, which will increase their desorption rate and will make it easier to identify them mass-spectrometrically.


References
- Gomberg, M., Chem. Ber., 1900, 33, 3150.
- Michelson, A.M.; Todd, A.R., J. Chem. Soc., 1953, 951.
- Smith, M., et al., J. Am. Chem. Soc., 1961, 84, 430.
- Greene, T.W.; Wuts, P.G.M., Protective Groups in Organic Synthesis, Wiley: USA, 1991.
- Fisher, E.F.; Caruthers, M.H., Nucleic Acids Res., 1983, 11, 1589.
- Vaughn-Settle, A., et al., Abstracts of papers of the ACS, 1997, 214, 102.
- Gildea, B.D., et al., Tetrahedron Lett., 1990, 31, 7095.
- Sekine, M.; Hata, T., J. Org. Chem., 1987, 52, 946.
- Chu, S.S.; Reich, S.H., Bioorg. Med. Chem. Lett., 1995, 5, 1053.
- Patel, V.F., et al., Bioorg. Med. Chem. Lett., 1995, 5, 513.
- Fourrey, J.L., et al., Tetrahedron Lett., 1987, 28, 5157.
- Shchepinov, M.S., et al., Nucl. Acids Res., 1997, 25, 1155.
- Smith, L.M., et al., Nature, 1986, 321, 674.
-
- Cardullo, F., et al., Proc. Natl. Acad. Sci. USA, 1988, 85, 8790-8794.
- Tyagi, S.; Kramer, F.R., Nature Biotech., 1996, 14, 303-308.
- Lewis, F.D., et al., J. Am. Chem. Soc., 1997, 119, 5451-5452.
- Masuko, M., et al., Nucl. Acids Symp. Ser., 1998, 39, 111-112. (e) Mao, C.D., et al., Nature, 1999, 397, 144-146.
- Dobrikov, M., et al., Nucleosides Nucleotides, 1999, 18, 1517-1518.
- Anderson, P.; Klewe, B., Acta Chem. Scand., 1967, 21, 2599.
- Shchepinov, M.S., et al., Tetrahedron Lett., 2000, 41, 4943.
- Klinger, K.W., et al., Cytogenetics and Cell Genetics, 1991, 58, 2149.
- Schena, M., et al., Science, 1995, 270, 467.
- Lee, L.G., et al., Nucleic Acids Res., 1997, 25, 2816.
-
- Eley, D.D.; Spivey, D.I., Faraday Soc. Trans., 1962, 58, 411.
- Lewis, F.D., et al., Science 1997, 277, 637.
- Wan, C., et al., Proc. Natl. Acad. Sci. USA, 1999, 96, 6014.
- Giese B., Chemistry in Britain, 2000, 44.
- Burroughes, J.N., et al., Nature, 1990, 347, 539.
- Whole issue, Curr. Opin. Chem. Biol., 1998, 2.
- Southern, E.M., et al., Genomics, 1992, 13, 1008.
- Furka, A., et al., 1988, Highlights of Modern Biochemistry, Proceedings of the 14th International Congress of Biochemistry VSP: Ultrecht, The Netherlands, 5, 47.
- Herz, M.L., J. Am. Chem. Soc., 1975, 97, 6777.
-
- Shchepinov, M.S.; Southern, E.M., Russ. J. Bioorg. Chem., 1998, 24, 794-797.
- Shchepinov, M.S., Glen Report, 1999, 12, 1.
- Shchepinov, M.S., et al., Tetrahedron, 2000, 56, 2713.
- Gioeli, C.; Chattopadhyaya, J.B., J. Chem. Soc., Chem. Commun., 1982, 672.