Glen Report 29.13: Studying DNA Repair Pathways Using Site-Specifically Modified Oligonucleotides
Young K. Cheun & Orlando D. Schärer
Center for Genomic Integrity
Institute for Basic Science
Ulsan National Institute of Science and Technology
Ulsan 44919, South Korea
young.cheun@ibs.re.kr
orlando.scharer@ibs.re.kr
1. Introduction
DNA is an inherently unstable molecule that can react with both endogenous (water, reactive oxygen species) and exogenous agents (UV light, environmental mutagens, antitumor agents).[1] Errors during replication of damaged sites in DNA leads to mutations in genes and eventually cancer. More recently, it has become clear that persistent DNA lesions are associated with many additional pathologies including premature aging and neurodegeneration.[2] Fortunately, a variety of pathways exist to repair DNA to counteract this threat. Mutations in DNA repair genes lead to debilitating inherited disorders and contribute to disease predisposition.
Conversely, DNA repair pathways have also become much sought-after targets for antitumor therapy for two main reasons.[3] First, a number of antitumor agents such as cisplatin or temozolomide cause cytotoxicity by inflicting DNA damage to tumor cells and the damage is processed by DNA repair enzymes similarly to endogenous DNA damage. The inhibition of DNA repair enzymes in tumors can therefore lead to improved therapeutic outcomes. Second, the vast majority of tumors have a defect in one of the DNA repair pathways. This allows them to acquire the number of mutations needed to develop into full-blown tumors. Such DNA repair defects provide specific vulnerabilities that can be exploited by targeting a second pathway that acts on the same type of lesion through the principle of synthetic lethality. This is the principle behind the clinically approved PARP inhibitor olaparib (trade name Lynparza), which was approved for the treatment of BRCA1/2-deficient cancers in 2015 and is on the way to become a blockbuster drug. Many start-up and established drug companies have programs for discovering the next wave of DNA repair inhibitors for use in oncology.
These developments were in no small part enabled by the detailed studies of the molecular mechanisms of DNA repair pathways, which in turn were made possible by using site-specifically modified oligonucleotides. We will discuss some examples of how the availability of phosphoramidites to synthesize oligonucleotides with DNA lesions has contributed to the field, using primarily structural studies for illustration.
2. DNA polymerases and glycosylases interacting with thymine glycol
Thymidine Glycol CE Phosphoramidite[4]
Thymine glycol (Tg, 5,6-dihydro-5,6-dihydroxythymine) is the most common oxidation product of thymine. It can be produced endogenously by aerobic metabolism or exogenously by chemical oxidants and ionizing radiation. The oxidation of the double bond in thymine leads to loss of aromaticity and base stacking, but does not affect the Watson-Crick base pairing properties. Tg is therefore not considered to be mutagenic, but it is a strong block to replication, making it a highly cytotoxic lesion.[5] Replication is blocked at the extension step immediately past the Tg rather than the insertion step opposite Tg. To explain this observation, Aller et al.[6] took a snapshot of the reaction of a replicative DNA polymerase with a Tg-containing oligonucleotide by X-ray crystallography. The structure offers insights into how Tg blocks DNA polymerase from extending beyond the lesion site (Figure 1). The loss of planarity of the pyrimidine ring places the methyl group into an axial position and induces steric hindrance forcing the 5' templating guanine out of the polymerase active site,[6]where it is stabilized in its misplaced position by the two vicinal diols of the Tg.[6] This guanosine is therefore unable to pair with dCTP, leading to a block of the polymerase reaction.
Figure 1: Thymine glycol (Tg) blocking replication |
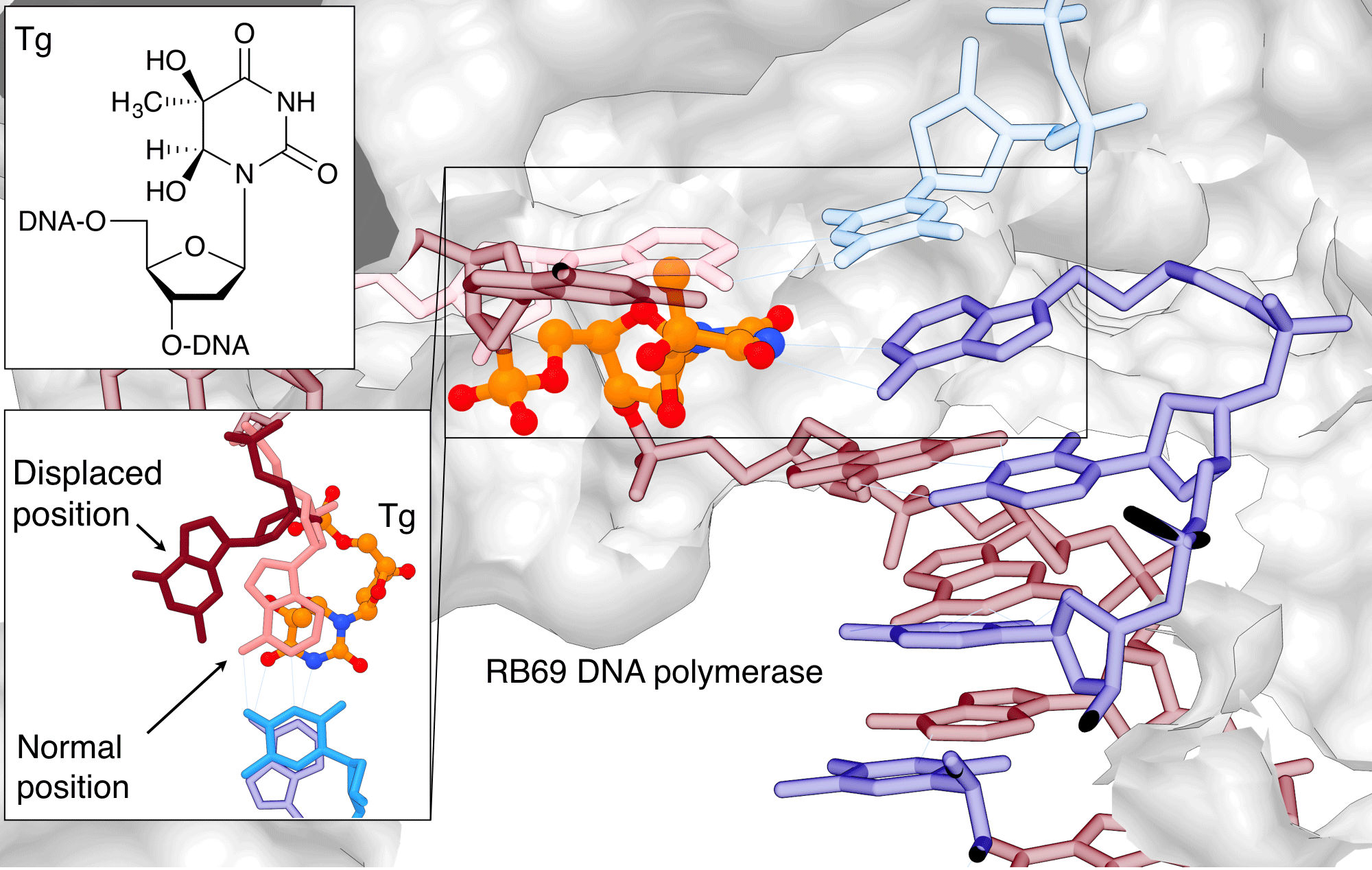 |
Thymine glycol (Tg) blocks the replication by displacing the base adjacent to the lesion so that it can no longer base pair during replication. Tg is shown in orange/atom color, the G adjacent to it in salmon (normal position) or burgundy (displaced position) The images were created using PDB ID 2DY4 and 1IG9 and UCSF Chimera. (See https://www.cgl.ucsf.edu/chimera/docs/licensing.html) |
Tg is primarily removed by base excision repair (BER) pathway. In BER, DNA glycosylases recognize specific lesions and cleave the glycosidic bond of the damaged base. The Neil family of DNA glycosylases cleave oxidized pyrimidines including Tg.[7] To gain information into this recognition process, Imamura et al.[8] used a Tg-containing oligonucleotide and a viral ortholog of NEIL1 glycosylase in their X-ray crystallographic studies. In the electron density map, the Tg lesion is flipped out of the helix to be positioned in the active site of the glycosylase (Figure 2).[8] This nucleotide flipping mechanism is conserved for most DNA glycosylases, allowing these enzymes to probe the modification outside the duplex, while gaining access to the glycosidic bond, which is normally hidden in the stack of the DNA duplex. Interestingly, NEIL1 makes few direct hydrogen bond interactions between the lesion and the amino acid residues in thymine glycol recognition site,[8] Instead, the propensity of Tg to assume an extrahelical conformation in DNA is a driving force for recognition by the Neil proteins.
Figure 2: MvNei1 forcing Thymine glycol (Tg) out of the helix |
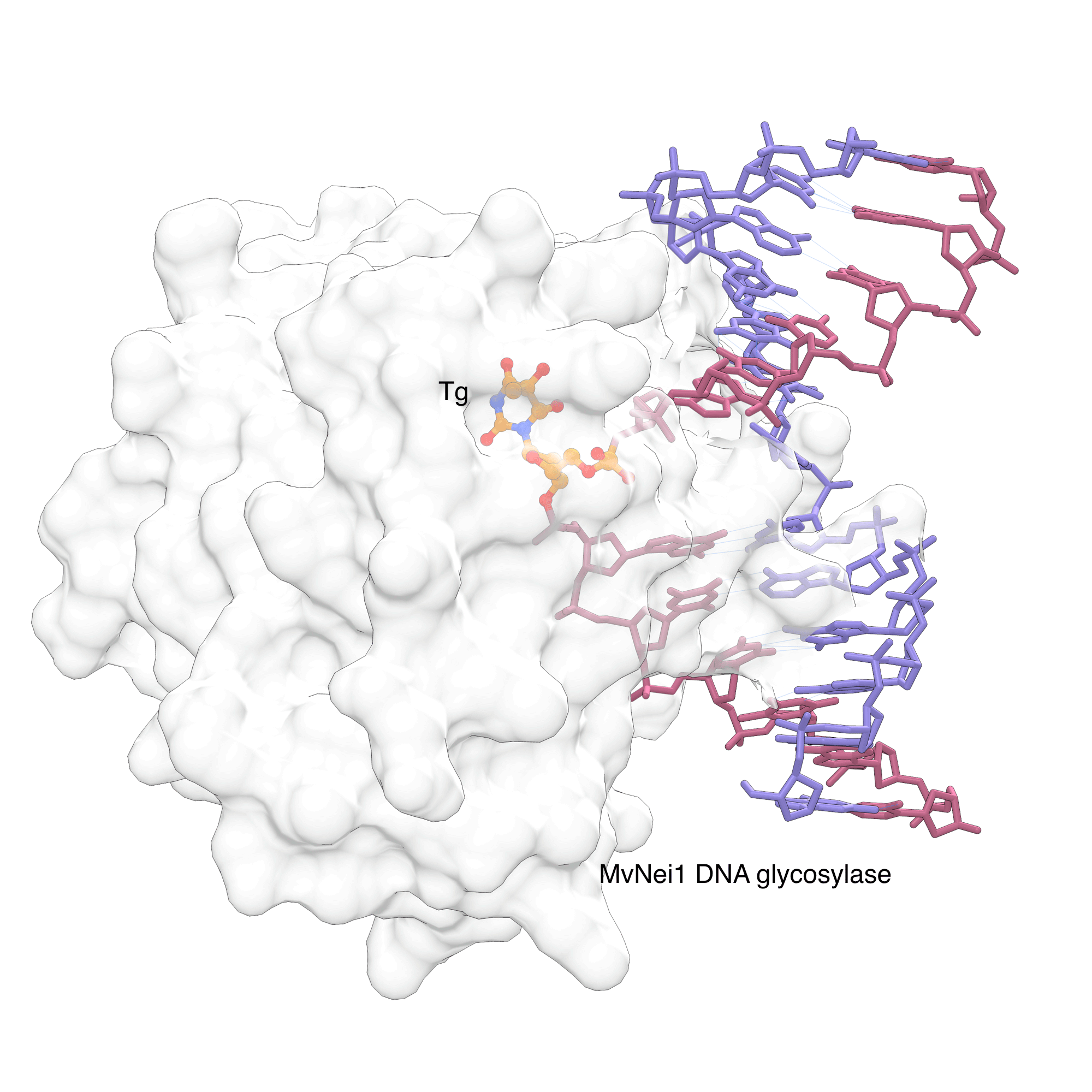 |
MvNei1, a viral ortholog of human DNA glycosylase NEIL1, can recognize the Tg lesion and force it to flip out of the helix. The flipped nucleotide is shown in orange/atom color in the base recognition site. The image was created using PDB ID 3VK8 and UCSF Chimera. |
Figure 3: hOGG1 discriminating 8-oxoG from G |
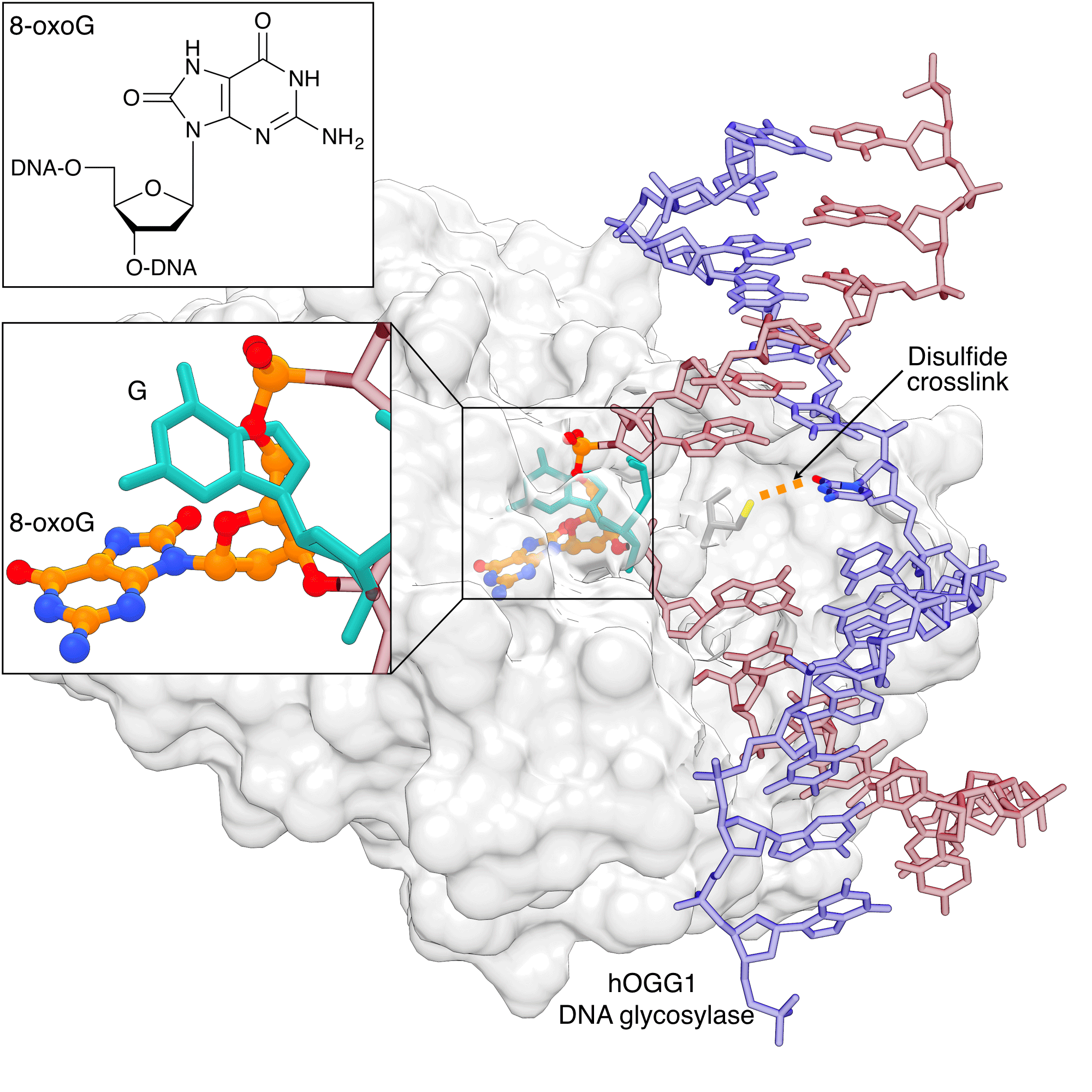 |
Human 8-oxoguanine DNA glycosylase I (hOGG1) can discriminate 8-oxoG from G before it is fully flipped out from the helix. The native (teal) and damaged (orange/atom color) bases are shown occupying distinct binding sites. The image was created by using PDB ID 1YQR and 1YQK and UCSF Chimera. |
3. Recognition of 8-Oxoguanine
8-oxo-dG-CE Phosphoramidite[9]
8-oxoguanine (8-oxoG) is the prototypical lesion caused by oxidative stress. Extensive studies of the recognition of 8-oxoG by the DNA glycosylase hOGG1 have not only revealed how this lesion is recognized in the flipped-out state, but also at various stages leading up to the ultimate recognition complex. Early structural studies showed early on that hOGG1 also recognizes DNA lesions by flipping out the 8-oxoG nucleotide into the active site pocket.[10] Dramatic insights into the how hOGG1 differentiates binding to 8-oxoG over non-damaged DNA were made possible through tethering the cysteine-engineered protein to the N4 cytosine of the DNA via a disulfide linkage using convertible nucleotide technology.[11] These studies showed that G and 8-oxoG occupy distinct sites on hOGG1 (Figure 3). These studies provided the basis for a full description of the recognition process of hOGG1 by a combination of structural, computational and biochemical studies.
4. Cyclobutane pyrimidine dimer (CPD) – the most important exogenous DNA adduct
Cis-syn Thymine Dimer phosphoramidite[12]
Cyclobutane pyrimidine dimers (CPDs) are perhaps the most important and well-known environmental DNA adducts. Under ultraviolet light (UV) exposure, two adjacent pyrimidines can undergo photochemical crosslinking reactions to form CPD (approx. 75%) and 6-4 pyrimidine-pyrimidone photoproducts (6-4 PPs, approx. 25%).[13] Like most bulky lesions, both are primarily repaired by nucleotide excision repair (NER). NER is a multistep process and includes two proteins that have affinity for bulky DNA adducts. XPC-RAD23B is a general damage sensor that recognizes lesions that thermodynamically destabilize the DNA duplex. UV-DDB is a more specialized protein complex that recognizes lesions in the context of chromatin. While UV-DDB is not required for the repair of all NER substrates, it is essential for NER of CPDs, because they induce very little helical distortion into a DNA duplex.[14]
The CPD phosphoramidite has allowed researchers to design an oligonucleotide with a CPD lesion at a specific position, playing a crucial role in elucidating the CPD repair mechanism by NER. The X-ray structure of UV-DDB–CPD-containing DNA by Fischer et al.[15] shows how the binding pocket of DDB2 subunit is optimized for the recognition of CPD lesions. It has a shallow binding pocket on the surface that can accommodate CPD and a wedge that helps extrude the CPD from the duplex (Figure 4). DDB2 is part of a ubiquitin ligase complex and its binding to DNA lesions triggers ubiquitination of DDB2 and XPC, facilitating binding XPC to damaged sites.
Min et al.[16] designed a "flipped-out CPD lesion as a substrate for Rad4-Rad23, the yeast ortholog of XPC-RAD23B, to illustrate the binding mode of the protein. Rad4 has two main DNA binding domains. The first one anchors the protein on the unmodified duplex DNA, and the other uses two beta-hairpins to encircle the unpaired bases on the non-damaged strand opposite the CPD lesion (Figure 4).[16] A more recent structure of Rad4 used convertible nucleotides to tether XPC to undamaged DNA, and showed that the binding mode is identical for damaged and undamaged DNA.[17] Together with spectroscopic and thermodynamic studies, the authors suggested that the binding affinity of Rad4 with DNA correlates with the propensity of a lesion to lead to an "open DNA structure, explaining the observed preference of NER to repair lesions that destabilize the DNA duplex. [17]
Figure 4: DDB2 recognizing the CPD lesion |
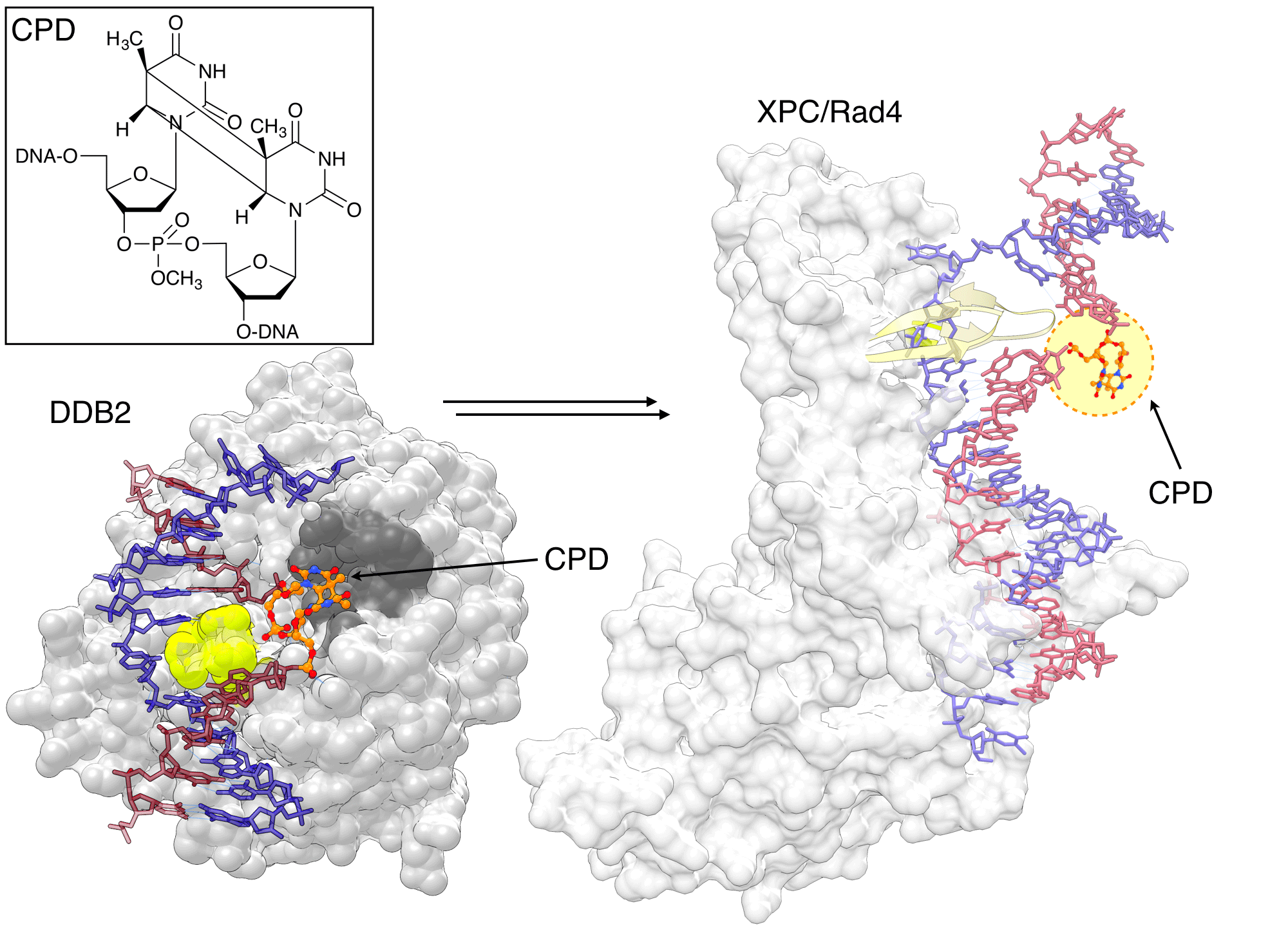 |
DDB2 is highly specialized for recognizing CPD lesion, which only mildly distort duplex. DDB2 has a shallow binding pocket (dark gray) to accommodate the CPD and a wedge (lime) to move it out of the duplex. Rad4/XPC binds to undamaged nucleotides opposite the lesion through two ß-hairpins (yellow) and does not contact the CPD (orange/atom color) directly. The CPD is not visible in the electron density map of the structure, but was modeled here for visual illustration. The image was created by using PDB ID 4A08 (left) and 2QSG (right) and UCSF Chimera. |
If a CPD lesion evades NER surveillance and the cell proceeds to the S phase, the bulky CPD can block replication. When a polymerase stalls at a lesion, a translesion synthesis (TLS) polymerase is recruited to bypass the lesion.[18] TLS polymerases have larger and more open active site cavities, allowing the bypass of DNA lesions, often at the expense of accuracy, in order to prevent the replication stalling. CPD-containing templates were used to solve X-ray structures of CPD lesion bound to Polη, showing how Polη can accurately bypass CPDs to prevent UV-induced mutations (Figure 5). The large active site of Polη can accommodate two bases of the CPD lesion in active site and use them as templating bases. Biertümpfel et al.[19] compared the X-ray structures of Polη in which the CPD lesion is located in either +1, +2, or +3 position after bypass. The authors found that the little finger (LF) domain, responsible for binding the template-primer, is very rigid and acts as a "molecular splint that straightens the CPD-containing DNA to maintain the shape of natural B-form, facilitating the correct insertions up to 3 base pairs after the CPD.[19] Consistent with biochemical studies, these structures suggested the CPD lesion is likely to experience a steric clash with the Polη after three bases[19], explaining the dissociation of Polη three bases after the CPD bypass.[20]
Figure 5: Human Polη bypassing CPD lesions |
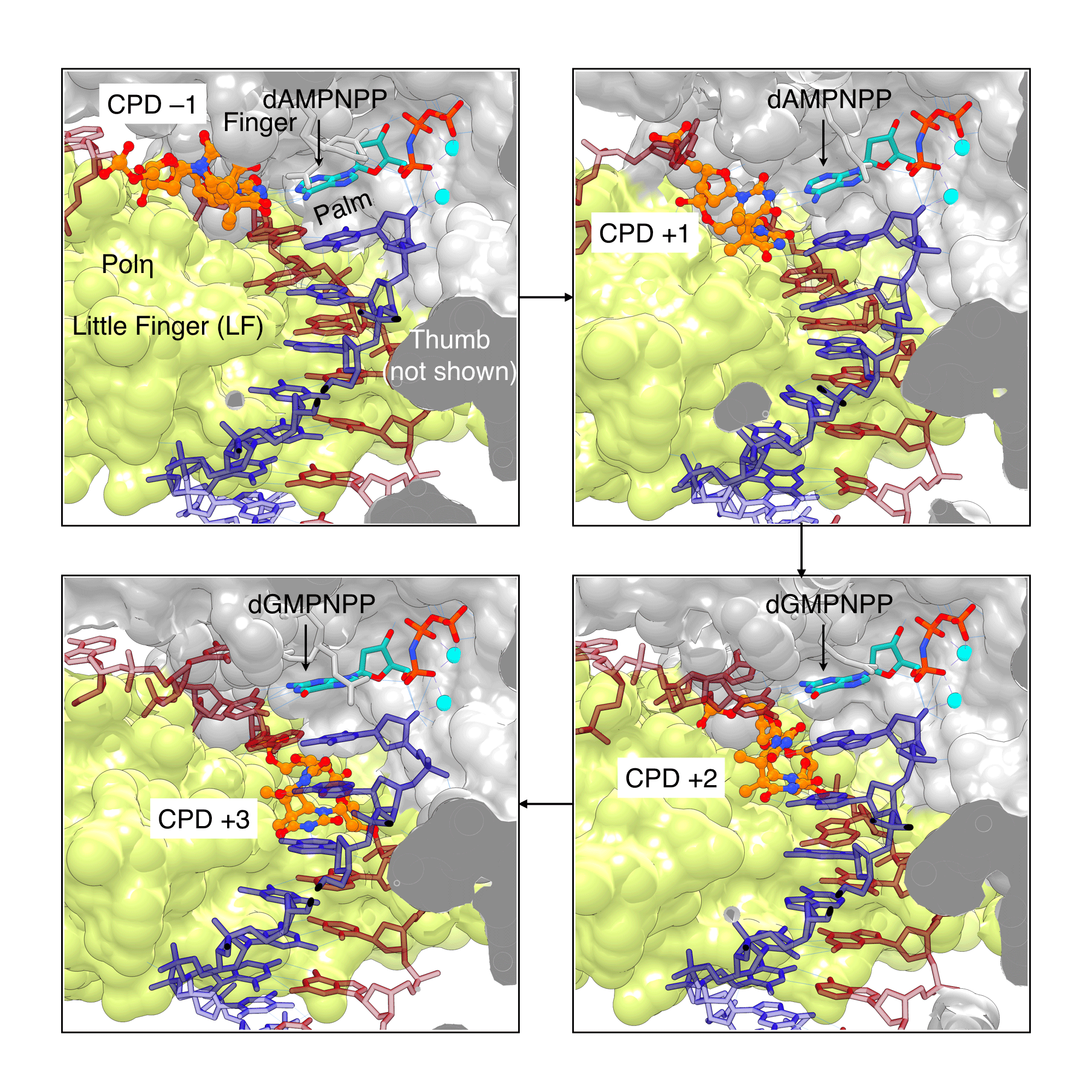 |
Human Polη is highly efficient at bypassing CPD lesions, as it can accommodate CPDs (orange/atom color) in its large active site and force the CPD-containing template into a “regular” B-DNA shape by using its little finger (LF) domain (lime) as a molecular splint. With the help of this splint, Polη can extend the primer 3 nucleotides past the lesion. The images were created using PDB ID 3MR3, 3SI8, 3MR5, and 3MR6 (clockwise from top left) and UCSF Chimera. |
5. Conclusions and Outlook
In summary, phosphoramidites allowing for the generation of oligonucleotides containing site-specific lesions have been vital components for studying the mechanism of DNA repair. For many research projects, obtaining the lesion-containing phosphoramidites is a limiting factor. New DNA lesions are still being discovered[21] and the study of their biological consequences will require their site-specific incorporation into oligonucleotides. In addition to some of the commercially available phosphoramidites discussed here, various oxidative DNA lesions, bulky DNA adducts formed by polyaromatic hydrocarbons or aromatic amines or the second major UV adduct – 6-4PPs are much sought-after lesions in studies of DNA repair enzymes. More complex lesions such as DNA interstrand crosslinks, which covalently link two strands of DNA and are formed by many antitumor agents[22] or DNA-protein crosslinks[23] have captured a lot of recent attention due to their dramatic effects on DNA replication. The increased availability of phosphoramidites for the synthesis of lesion-containing oligonucleotides should facilitate many future discoveries in the broad area of DNA damage and repair.
References
[1] T. Lindahl, Angew. Chem. Int. Ed. 2016, 55, 8528-8534.
[2] J. H. Hoeijmakers, N. Engl. J. Med. 2009, 361, 1475-1485.
[3] M. J. O'Connor, Mol. Cell 2015, 60, 547-560.
[4] S. Iwai, Angew. Chem. Int. Ed. 2000, 39, 3874-3876.
[5] J. M. Clark, G. P. Beardsley, Nucleic Acids Res. 1986, 14, 737-749.
[6] P. Aller, M. A. Rould, M. Hogg, S. S. Wallace, S. Doublié, Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 814-818.
[7] R. P. Hickerson, C. L. Chepanoske, S. D. Williams, S. S. David, C. J. Burrows, J. Am. Chem. Soc. 1999, 121, 9901-9902.
[8] K. Imamura, A. Averill, S. S. Wallace, S. Doublié, J. Biol. Chem. 2012, 287, 4288-4298.
[9] V. Bodepudi, S. Shibutani, F. Johnson, Chem. Res. Toxicol. 1992, 5, 608-617.
[10] S. D. Bruner, D. P. G. Norman, G. L. Verdine, Nature 2000, 403, 859-866.
[11] A. Banerjee, W. Yang, M. Karplus, G. L. Verdine, Nature 2005, 434, 612-618.
[12] aJ. S. Taylor, I. R. Brockie, C. L. O'day, J. Am. Chem. Soc. 1987, 109, 6735-6742; bT. Murata, S. Iwai, E. Ohtsuka, Nucleic Acids Res. 1990, 18, 7279-7286.
[13] D. L. Mitchell, G. M. Adair, R. S. Nairn, Photochem. Photobiol. 1989, 50, 639-646.
[14] O. D. Schärer, Cold Spring Harb. Perspect. Biol. 2013, 5, 1-19.
[15] E. S. Fischer, A. Scrima, K. Bohm, S. Matsumoto, G. M. Lingaraju, M. Faty, T. Yasuda, S. Cavadini, M. Wakasugi, F. Hanaoka, S. Iwai, H. Gut, K. Sugasawa, N. H. Thomä, Cell 2011, 147, 1024-1039.
[16] J. H. Min, N. P. Pavletich, Nature 2007, 449, 570-575.
[17] X. J. Chen, Y. Velmurugu, G. Q. Zheng, B. Park, Y. Shim, Y. Kim, L. L. Liu, B. Van Houten, C. He, A. Ansari, J. H. Min, Nat. Commun. 2015, 6, 1-10.
[18] S. S. Lange, K. Takata, R. D. Wood, Nat. Rev. Cancer 2011, 11, 96-110.
[19] C. Biertümpfel, Y. Zhao, Y. Kondo, S. Ramón-Maiques, M. Gregory, J. Y. Lee, C. Masutani, A. R. Lehmann, F. Hanaoka, W. Yang, Nature 2011, 476, 360-360.
[20] S. D. McCulloch, R. J. Kokoska, C. Masutani, S. Iwai, F. Hanaoka, T. A. Kunkel, Nature 2004, 428, 97-100.
[21] Y. Yu, P. Wang, Y. Cui, Y. Wang, Anal. Chem. 2017,
[22] C. Clauson, O. D. Schärer, L. Niedernhofer, Cold Spring Harb. Perspect. Biol. 2013, 5, 1–25.
[23] N. Y. Tretyakova, A. t. Groehler, S. Ji, Acc. Chem. Res. 2015, 48, 1631-1644.
Product Information
Thymidine Glycol CE Phosphoramidite (10-1096)