Glen Report 28.14: New Product - Depurination Resistant dA-CE Phosphoramidite
Introduction
Depurination is defined as the cleavage of the glycosidic bond attaching a purine base to the sugar moiety. Electron withdrawing acyl protecting groups like benzoyl and isobutyryl on the purine amino group(s) destabilize the glycosidic bond, whereas electron donating formamidine protecting groups stabilize the glycosidic bond. The use of formamidine protecting groups to eliminate depurination was first proposed1,2 in the 1980s and dmf-dG is an example of this strategy in routine use today.
The consequence of depurination during oligonucleotide synthesis is the loss of the purine base to form an internucleotide linkage containing the abasic sugar at that position. This site is stable during further synthesis cycles but, upon deprotection with basic reagents, the oligonucleotide is cleaved at that position leading to two shorter fragments. The fragment towards the 5’ terminus still contains the DMT group. If DMT-ON purification is being used, the depurinated fragments are co-purified along with the full length product as truncated oligonucleotides.
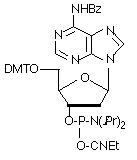
dA is actually more susceptible to depurination than dG and the dA monomer most commonly used is protected with the destabilizing benzoyl group (1). So why has this not been a factor in routine oligonucleotide synthesis? Undoubtedly, depurination does occur in regular oligonucleotide synthesis but at an extremely low level due to the exquisite balancing of the steps in the synthesis cycle. Nevertheless, our data show that if a dA20dT oligo is tortured by changing the acid deprotection step to 15 minutes per cycle, then the oligo is almost completely degraded and only a small amount of full length product is observed.
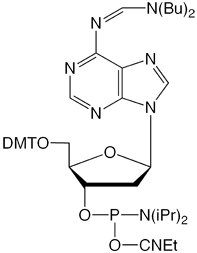
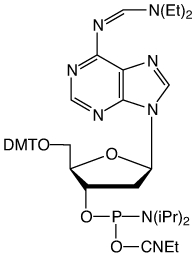
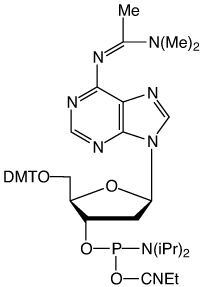
Figure 1: Structures of protected dA-CE Phosphoramidtes |
|||
 |
 |
 |
 |
bz-dA (10-1000) (1) |
dbf-dA (10-1500) (2) |
def-dA (10-1504) (3) |
dma-dA (10-1505) (4) |
Figure 2: RP HPLC of DMT-ON dT Oligo Containing 3 Insertions of dma-dA

Figure 3: IEX HPLC of Protected dA10dT Tortured with TCA

Depurination may not be very significant in routine synthesis but it is still a problem in certain other circumstances, as noted below:
Synthesis of long oligos3
In this scenario, the exposure of protected dA sites to acid during the deprotection step, although brief each cycle, accumulates as the synthesis proceeds. Trichloroacetic acid (TCA), which is the standard acid used in routine small scale oligo synthesis, is quite strong, with a pKa of approximately 0.7. For the synthesis of long oligos, an alternative is to use a deblocking agent with a higher pKa. The most common choice is dichloroacetic acid (DCA), which has a pKa of 1.5. However, the risk of depurination using TCA must be balanced with the risk of incomplete deprotection using DCA. Depurination may lead to shorter fragments truncated at the 3’ terminus but incomplete deprotection would lead to deletion mutations in a family of n-1, n-2, etc., oligos and this may be an even worse scenario for such applications as gene construction. This situation would benefit from a depurination resistant dA monomer.
Chip-Based Synthesis
The synthesis of microarrays on silicon wafers requires the synthesis methodology that works so well on the 3-dimensional porous surface of CPG to be adapted to a 2-dimensional non-porous surface. If the synthesis strategy requires acid to be used for detritylation, there is the potential for surface effects to cause increased concentration of the acid, potentially leading to increased levels of depurination. Indeed, this very situation on Agilent’s DNA microarray synthesis platform led to the conclusion that depurination is the limiting factor in the ability to synthesize long oligos (up to 150mers) – even using DCA as the detritylating reagent.4 In this case, depurination had to be controlled by adjusting the fluidics of the flow cell and by quenching the detritylation solution with oxidizer solution. A depurination resistant dA monomer may also significantly improve this situation.
Large-Scale Synthesis
The extended reaction times of large-scale synthesis require careful consideration of depurination potential. On the other hand, cycle optimization has been so successful and costs are so tightly controlled that a more expensive depurination resistant dA monomer may well have been rejected previously.
Depurination Resistant dA
We have discussed this topic in the past while introducing the dbf-dA monomer (2). This dibutylformamidine protected dA seemed a likely candidate for the synthesis of long oligos but it did not perform as well as we expected and was discontinued. We surmise that the problem occurred during deprotection when the dibutylformamidine group may have been initially hydrolyzed to form dibutylamine, which is both involatile and capable of forming a salt with the phosphate backbone.
In response, we have gone back to basics and prepared the diethylformamidine (def) (3) and the dimethylacetamidine (dma) (4) protected dA monomers. In the former case, diethylamine would be formed on deprotection and this is highly volatile. In the latter case, dimethylacetamide is fairly involatile but would be unlikely to form a salt with the phosphate backbone.
Rate of deprotection
In the first instance, we looked at the relative rates of deprotection and they are collected in Table 1. As expected, def-dA was rapidly deprotected in ammonium hydroxide and AMA. Surprisingly, we found that dma-dA was deprotected quickly in ammonium hydroxide but it took 80 minutes at 65° in AMA for complete deprotection. The time course of this deprotection is shown in Figure 2.
Table 1: Time Required for Complete Deprotection |
||
Protecting group
|
30% Ammonium
|
Ammonium Hydroxide/
|
|
Benzoyl |
<2 hours |
<10 minutes |
|
dibutylformamidine |
2 hours |
20 minutes |
|
diethylformamidine |
2 hours |
20 minutes |
|
dimethylacetamidine |
4 hours |
80 minutes |
Depurination Resistance
As a further experiment, we carried out a torture synthesis of dA10dT using 3% TCA/DCM and with detritylation steps increased to 15 minutes to simulate the synthesis of an oligo approximately 15 times longer. The resulting oligonucleotides were purified DMT-ON with GlenPak cartridges to remove any oligonucleotides that did not contain 5'-DMT species. The resulting oligonucleotides were then analyzed by ion exchange HPLC, as shown in Figure 3.
In the case of Bz-protected dA, the oligonucleotide was substantially degraded. However, in the case of both of these new monomers, the oligonucleotide was well protected against depurination and a good yield of full length product was achieved.
Conclusion
In this article, we have demonstrated that the most commonly used dA-CE Phosphoramidite containing benzoyl protecting groups suffers substantial degradation by depurination after excessive exposure to TCA. However, the TCA exposure in our torture treatment of dA10dT (15 minutes per cycle or 150 minutes total) is equivalent to the exposure experienced by a dA residue adjacent to the 3' terminus during the synthesis of a 150mer on the LV200 cycle of an ABI synthesizer.
At the same time, we have demonstrated that two depurination resistant dA monomers, protected with diethyl-formamidine (def) and dimethylacetamidine (dma), are essentially stable to depurination during the torture testing.
Testing of the rates of deprotection of the two new monomers showed that both were rapidly deprotected in ammonium hydroxide and are fully compatible with regular deprotection strategies. However, an interesting anomaly was observed during deprotection with popular AMA at 65°. Def-protected-dA was rapidly deprotected in 20 minutes, which makes it fully compatible with regular AMA deprotection. In contrast, the dma-protected-dA was only about 50% deprotected by AMA in 20 minutes at 65°. A time course study revealed that 80 minutes were required for complete deprotection.
Due to the longer time of 80 minutes required for complete deprotection of dma-dA with AMA, we have decided to offer the def-dA for routine synthesis requiring depurination resistance. We leave open the option of supplying dma-dA for more specific applications.
References
1. B.C. Froehler, and M.D. Matteucci, Nucleic Acids Res, 1983, 11, 8031-6.
2. L.J. McBride, R. Kierzek, S.L. Beaucage, and M.H. Caruthers, J. Amer. Chem. Soc., 1986, 108, 2040.
3. The Glen Report, 2009, 21.2, 14-15.
4. E.M. LeProust, et al., Nucl Acid Res, 2010, 38, 2522-2540.