
For the last four years, we have been aware of the need for suitable solid-phase synthesis procedures to supplement the enzymatic techniques used to produce oligoribonucleotides. We have watched with interest as RNA phosphoramidites became commercially available but were disappointed to hear of problems encountered by end-users - cloudy monomer solutions , low monomer purities, and low coupling efficiencies - which, in some cases, led to some disappointing results. We do not make these comments simply to be critical of current suppliers - quite the contrary. The scale-up of the synthesis of RNA phosphoramidites is truly complex and it is to the great credit of the manufacturers that supplies have been maintained. Indeed, we know, to our cost, just how difficult the processes are! So, what are the problems in the production of RNA monomers and in automated RNA synthesis?
The only difference in a ribonucleoside relative to the corresponding 2'-deoxyribonucleoside is, obviously, the presence of the 2'-hydroxyl group. What can possibly happen at the 2' position 1 to make RNA synthesis so much more complex than DNA synthesis? First of all, a protecting group must be selected for the 2' position which will allow the preparation of the phosphoramidite monomer without migration of the 2' protecting group to the 3' position. Assuming this can be achieved, the protecting group must be stable to the conditions of automated synthesis without its loss or migration. Indeed, if it is lost during synthesis, chain scisslon may occur at the adjacent phosphodiester linkage during the ammonium hydroxide removal of the base protecting groups. Finally, the protecting group must be efficiently removed under mild conditions to generate the product ollgoribonucleotide.
Most synthetic strategies have been designed to carry out automated DNA and RNA synthesis in the 3' to 5' direction. The 5'-hydroxyl group is usually protected with the 4,4'-dimethoxytrityl (DMT) group which is labile to dilute organic acid and generates a colored cation which can be monitored to give a measure of synthesis efficiency. While the combination of acid-labile DMT groups at the 5' position and base-labile protecting groups on the bases, as used routinely for DNA synthesis, is very convenient, it places severe constraints on the nature of the group used for 2' protection.
Protecting groups which are stable in the presence of acid and base are clearly prime candidates for use in 2' protection. For example, the 2-nitrobenzyl group2 can be removed photochemically and the 3,4- dimethoxybenzyl3 group can be oxidatively cleaved. However 1 in both cases, incomplete deblocking has been observed in the synthesis of long RNA fragments. The t-butyldimethyl silyl (TBDMS) group, extensively investigated by Ogilvie and coworkers 4 over many years, is also acid and base stable and can be removed relatively conveniently with fluoride ion. Indeed, 2'-O-TBDMS protection is the basis for all RNA monomers which are currently commercially available. The 2'-acetal protecting groups, tetrahydropyranyl {THP) and methoxytetrahydropyranyl (MTHP), are cleaved under aqueous conditions at pH 2 . As such, these groups are potentially very convenient since they allow a simple deprotection and work up just prior to use of the synthetic RNA fragments . However, both groups have proved to be too unstable5 to acid to survive many detritylation cycles.
Protection of RNA monomers with 2'-O-MTHP in conjunction with base-labile 9-fluorenylmethoxycarbonyl (Fmoc) protection of the 5'-hydroxyl groups offers an interesting alternative synthetic strategy6. The coupling efficiency per cycle could still be monitored by measuring the release of the Fmoc group spectrophotometrically. Monomers for this approach have not yet been made available commercially.
Further refinements of 2'-acetal protection strategies have led to the development of 1-(2-chloro-4- methylphenyl)-4-methoxypiperidin-4-yl (CTMP) 1 and 1-(2-fluorophenyl)-4 - methoxypiperidin-4-yl (FPMP) by Reese and coworkers. These 2' protecting groups are unaffected by DCA deprotectlon during the normal synthesis cycles, but can be conveniently removed at pH 2 just prior to the use of the synthetic RNA fragment. Other 2'-acetal protecting groups which have been evaluated include 3-methoxy-1,5-dicarbo-methoxypentyl8 and 1-(2-chloroethoxy) ethyl9. It is disappointing that monomers using this approach have not been made available commercially since this protection scheme seems to be close to optimal for automated RNA synthesis and facile isolation of the product RNA with reduced risk of RNase degradation. A recent paper by Sproat and coworkers 10 provides an excellent overview of the state of play in 2' protection, while opting for the use of FPMP.
While the merits and demerits of various protection schemes can be debated at length, there is little doubt that most experience in RNA synthesis has been gained using the 2'-O-TBDMS protected monomers, described by Ogilvie and coworkers, since these have been commercially available for some time . The main problem with the use of these monomers for RNA synthesis has been the partial loss of the 2' protecting group leading to chain scission during the extended ammonium hydroxide deprotection necessary tor removal of the standard base protecting groups . These problems have been largely overcome11 by the use of base-labile protecting groups similar to those proposed for DNA synthesis12.
At this point in the development of automated RNA synthesis, the combination of highly base-labile protecting groups along wlth 2'-O-TBDMS protection seems to us to offer the best approach currently available. Glen Research is happy to offer monomers based on this approach to RNA synthesis (Figure 1). Moreover, we are committed to providing only monomers which adhere to strict performance criteria, including >97% coupling and >97% purily by HPLC. We guarantee that we will adhere strictly to these quality specifications to avoid wasting researchers' time wrestling with poor quality products .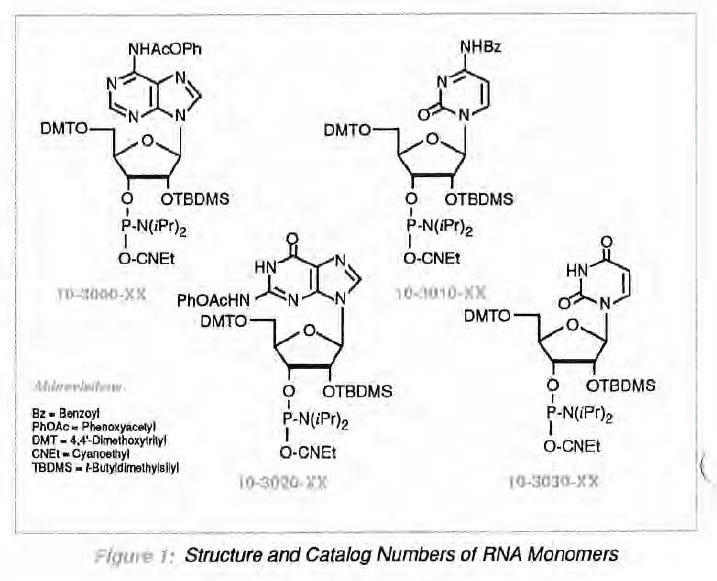
Having carried out automated synthesis of oligoribonuc!eotides, the researcher is faced with the following difficulties.
1) How are the base protecting groups to be removed without affecting the 2' protecting groups? Remember, if the 2' protecting group is lost, the ammonium hydroxide, necessary to remove the base protecting groups, may cause chain cleavage at the adjacent phosphodlester linkage.
2) How are the 2' protecting groups removed quantitatively to give biologically active RNA?
3) What are the options for purification of the synthetic RNA?
Assuming the use of good quality monomers, RNA synthesis proceeds in a manner virtually identical to its DNA counterpart, the exception being the coupling time which is extended to 15 minutes to compensate for the presence of the bulky 2' protecting group. It is possible to use a more acidic activator, e.g., 4-nitrophenyl tetrazole10, to speed up the coupling step and we will be examining a selection of these in the coming months. However, changes in the normal synthetic DNA deprotectlon scheme become essential.
Using standard base protecting groups and 2'-O-t-butyldimethylsilyl (TBDMS) protection, it was found13 that the standard ammonia solution used for deprotection in DNA synthesis has to be modified for RNA synthesis because aqueous ammonia solutions cause significant loss of the silyl protecting groups which, in turn, may lead to chain scission . When the ammonia solution was modified by using a mixture of ammonium hydroxide - ethanol (3:1) for 17 hours at 55°C chain cleavage was lowered to less than 1%. However, the authors note that the use of more base-labile protecting groups would be advantageous . The harsh conditions involving the use of aqueous ethanolic ammonium hydroxide can be moderated if base-labile protecting groups are used to replace the groups normally used (isobutyryl for G, and benzoyl for A and C). Phenoxyacetyl protection for A and G has been described12 for use in DNA synthesis. This protection scheme, along with similar base-labile protection for C, was evaluated by Ogilvie and coworkers11. Using ammonium hydroxide-ethanol (3:1) at room temperature, it was found that desilylation could be markedly reduced, although it was still observed. Also, the C monomer was found to be sparingly soluble in acetonitrile, and so, N-benzoyl-C was used for further studies. The authors then evaluated the use of anhydrous methanolic ammonia for base deprotection. It was found that deacylation proceeded rapidly (8 hours) at room temperature and completely eliminated desilylation and resulting chain scission.
A recent report14 extends the use of anhydrous alcoholic ammonia to oligoribonucleotides containing the standard (isobutyryl for G, and benzoyl for A and C) base protecting groups. Ethanolic ammonia, being less volatile than methanolic ammonia, was used for deprotection at elevated temperatures (16 hours at 55°C). Again, no silyl loss was observed.
The removal of the TBDMS group rrom the 2' position is accomplished11 without phosphate migration or cleavage using 1M tetrabutylammonium fluoride (TBAF) in THF (24 hours at room temperature). The excess TBAF is normally removed by desalting, but a simple procedure using an ion exchange cartridge has also been described14.
Polyacrylamide gel electrophoresis (PAGE) is the method of choice for purification of synthetic RNA. Elution from the gel is followed by desalting and lyophilization of the product. Purification by ion exchange or reverse phase (RP) HPLC has been described less frequently, presumably because of the danger of ribonuclease contamination in these procedures. RP cartridge purification would clearly have similar drawbacks. Analysis of synthetic RNA can be carried out using PAGE but rigorous confirmation that the product contains only 3'-5' linkages (rather than 2'-5' linkages produced by migration before, during or after synthesis) requires phosphodiesterase digestion of a sample of the product, followed by HPLC analysis of the nucleotides formed. Having made it this far, how do you avoid losing the precious synthetic RNA to ubiquitous ribonuclease enzymes? Once the 2' protecting groups have been removed, RNA must be worked with under scrupulously sterile conditions. Details of acceptable sterilization procedure and handling techniques are contained in standard molecular biology texts.
(1) C.B. Reese, Nuc/eos/das & Nucleotides, 1987, 6, 121-129.
(2) T. Tanaka, S. Tamatsukuri, and M. Ikehara, Nucleic Acids Res., 1987, 15, 7235.
(3) H. Takaku, T. Ito, and K. Imai, Chemistry Letters , 1986, 1005.
(4) 1. Wu and K.K. Ogilvie, J. Org. Chem., 1990, 55, 4717, and references cited therein.
(5) C. Christodoulou , S. Agrawal, and M.J. Gait, Tetrahedron Lett., 1986, 27, 1521.
(6) C. Lehmann, Y.Z. Xu, C. Christodoulou, Z.K. Tan, and M.J. Gait, NuclaicAcids Res., 1989, 17, 2379 .
(7) C.B. Reese and E.A. Thompson, J. Chem. Soc. Perkin Trans. t, 1988, 2881.
(8) A. Sandstrom, M. Kwiatkowski, and J.B. Chattopadhayaya, Acta Chem. Scand., 1985, B39, 273.
(9) S. Yamakage, 0 . Sakatsume, E. Furuyama, and H. Takaku , Tetrahedron Lett., 1989, 30, 6361.
(10) B. Beijer, I. Sulston, B.S. Sproat, P. Ridet, A.I. Lamond, and P. Neuner, Nuclefc Acids Res., 1990, 18, 5143-5151.
(11) T. Wu, K.K. Ogilvie, and R.T. Po11N, ucleic Acids Res., 1989, 17, 3501.
(12) J.C. Schulhof, D. Molko, and R. Teoule, Nucleic Acids Res., 1987, 15,397.
(13) J. Stawinski, R. Stromberg, M. Thelin , and E. Westman , Nucleic Acids Res,, 1988, 16, 9285.
(14) S.A. Scaringe , C. Francklyn, and N. Usman, Nuc/efc Acids Res., 1990, 18, 5433-5441 ,