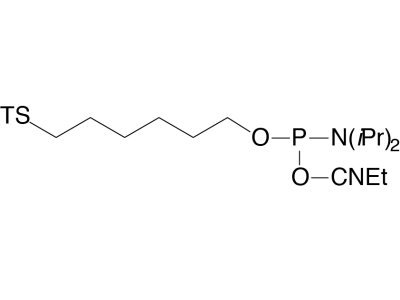
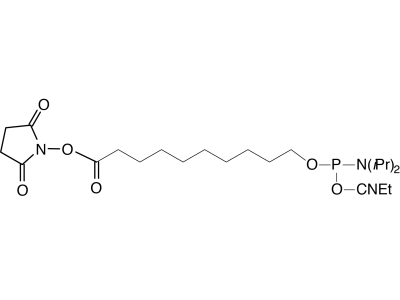
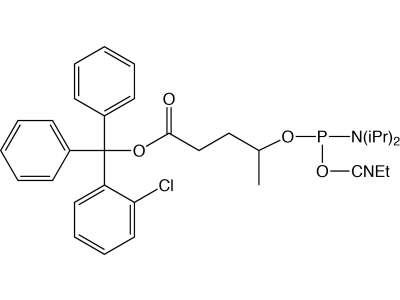
The disulfide thiol modifier may be used for introducing 3’- or 5’-thiol linkages. Dithiol Serinol, produced from lipoic acid and our patented serinol backbone, allows easy connection of multiple dithiol-labelled oligos to gold surfaces. 5’-Carboxy-Modifier C10 is a unique linker designed to be added at the terminus of an oligonucleotide synthesis. It generates an activated carboxylic acid N-hydroxysuccinimide (NHS) ester suitable for immediate conjugation on the synthesis column with molecules containing a primary amine, resulting in a stable amide linkage. An alternative carboxylate protecting group is the 2-chlorotrityl group, which is simply removed using the standard deblock cycle to generate a free carboxyl group on an otherwise fully protected oligonucleotide. The 2-chlorotrityl group is also removed during oligo deprotection with ammonium hydroxide or AMA and is incompatible with RP purification techniques. PC Amino-Modifier is a photocleavable C6 amino-modifier, part of our line of photocleavable (PC) modifiers. 5’-AminoOxy-Modifier 11 is based on a tetraethylene glycol linkage for improved solubility and for reducing the potential negative impact on hybridization of the oligo. The oxime formed from the reaction of alkyloxyamines with aldehydes creates a stable covalent bond. In comparison, the imine formed by the conjugation of primary amines with aldehydes is not stable to acidic or basic conditions and requires subsequent reduction with borohydride to form stable amine conjugates. 5’-Maleimide Modifier Phosphoramidite, developed at the University of Barcelona, incorporates a maleimide cycloadduct that is stable to ammonium hydroxide at room temperature. This phosphoramidite can be incorporated into DNA and RNA with both phosphate and phosphorothioate linkages. A retro–Diels-Alder reaction deprotects the maleimide immediately prior to conjugation.
Details
Usage
Coupling: Standard coupling time. Use 0.02 M Iodine for Oxidation.
Deprotection: After normal deprotection, the disulfide can be cleaved at room temperature in 30 minutes with 100 mM DTT pH 8.3 - 8.5 in the buffer of your choice.
Specifications
Diluent
Anhydrous Acetonitrile
Storage
Freezer storage, -10 to -30°C, dry
Stability
2-3 days
Dilution/Coupling Data
The table below show pack size data and, for solutions, dilution and approximate coupling based on normal priming procedures.
No. However, this can be produced on the synthesizer by adding to the 5'- terminus first 5'-thiol-modifier C6 S-S (10-1936) followed by BioTEG phosphoramidite (10-1955). This should generate a biotinylated primer with a long spacer arm containing the disulfide linkage which can be cleaved later with DTT.||
It would seem that the best method to make peptide-oligo conjugates would be to use Fmoc chemistry and synthesize the peptide off an oligo synthesized on amino-CPG. However, deprotection of peptides synthesized using Fmoc chemistry requires 50% TFA and t-boc synthesized peptides require HF both of which would severely damage if not completely hydrolyze the oligo.The best and most straight foward method is to use a heterobifunctional crosslinking reagent to link a synthetic peptide, containing an N-terminal lysine, to a 5'-Thiol modified oligo or conversely a 5'-amino modified oligo to a cysteine containing peptide . A good crosslinking reagent is N-Maleimido-6-aminocaproyl- (2'-nitro,4'-sulfonic acid)-phenyl ester . Na + (mal-sac-HNSA) from Bachem Bioscience (cat. # Q-1615). Reaction of this crosslinker with an amino group releases the dianion phenolate, 1-hydroxy-2-nitro -4-benzene sulfonic acid a yellow chromophore. The chromophore allows both quantitation of the coupling reaction as well as act as an aid in monitoring the seperation of "activated peptide" from free crosslinking reagent using gel filtration. Method A: Couple Peptide Amine To Oligo Thiol (Note peptide MW must be > 5,000 to be excluded from desalting column). This method best for oligo-enzyme conjugation. Step 1: Synthesize a peptide with an N-terminal, or internal, lysine (The epsilon amino group is more reactive than an alpha amino group). Step 2: Synthesize an oligonucleotide with a 5' Thiol group. Step 3: React peptide with excess mal-sac-HNSA (pH 7.5 Sodium phosphate) Step 4: Seperation of peptide-mal-sac conjugate from free crosslinker and buffer exchange (pH 6.0 Sodium phosphate) using a gel filtration column (Glen Gel-Pak™ or eq.). Note peptide must be large enough to seperate from the free linker which can be visualized as a yellow band. Do not collect yellow band with peptide. Step 5: Activate thiol modified oligo, desalt and buffer exchange (pH 6 Sodium phosphate) on Glen Gel-Pak™ column. Step 6: React acitvated peptide with Thiol modified oligo. Step 7: Purify Peptide-Oligo conjugate by ion exchange chromatography on Nucleogen DEAE-500-10 or eq. Elution order: free peptide, peptide-oligo, free oligo. Method B: Couple Oligo Amine To Peptide Cysteine (Note oligos > 15mers are excluded from desalting column). Use above procedure switching oligo for peptide. Step 1: Synthesize a peptide with an N-terminal, or internal, cysteine Step 2: Synthesize an oligonucleotide with a 5' amino modifier. Step 3: Purify oligo Trityl-on by RP HPLC or cartridge. Step 4: React oligo with excess mal-sac-HNSA (pH 7.5 Sodium phosphate) Step 5: Seperation of oligo-mal-sac conjugate from free crosslinker and buffer exchange (pH 6 Sodium phosphate) using a gel filtration column (Glen Gel-Pak™ or eq.). Note oligo must be large enough to seperate from the free linker which can be visualized as a yellow band. Do not collect yellow band with oligo. Step 6: Dissolve peptide in pH 6.0 Sodium phosphate buffer and react with activated oligo. Step 7: Purify Peptide-Oligo conjugate by ion exchange chromatography on Nucleogen DEAE-500-10 or eq. Elution order: free peptide, peptide-oligo, free oligo.